You might also like
- Primeros AuxiliosDocument33 pagesPrimeros AuxiliosLuOnofreHuamani100% (1)
- 7.exposición BPM GALENICOSDocument27 pages7.exposición BPM GALENICOSIsabel SeguraNo ratings yet
- Registro Sanitario1Document16 pagesRegistro Sanitario1galacmanNo ratings yet
- Vías de AdministraciónDocument30 pagesVías de AdministraciónツImSonnyNo ratings yet
- Apuntes de Alquimia TaoístaDocument51 pagesApuntes de Alquimia Taoístam3rl33nuxNo ratings yet
- Sistema Peruano de FarmacovigilanciaDocument48 pagesSistema Peruano de FarmacovigilanciaDebora Espinoza100% (1)
- Codigo ATC 123Document103 pagesCodigo ATC 123Alonso SardónNo ratings yet
- Generalidades Del PetitorioDocument22 pagesGeneralidades Del PetitorioArturo Irvin Beltran RiosNo ratings yet
- Expo Cajabamba 2022Document43 pagesExpo Cajabamba 2022Edgar Percy Burgos ReynaNo ratings yet
- Sistema Peruano Farmacovigilancia TecnovigilanciaDocument49 pagesSistema Peruano Farmacovigilancia TecnovigilanciaCINTHYA MASSIEL CABRERA QUEVEDO100% (1)
- 1 Clase Aspectos Legales en La Industria Farmaceutica - 5 CicloDocument28 pages1 Clase Aspectos Legales en La Industria Farmaceutica - 5 CicloDante RamosNo ratings yet
- Adquisicion de Los Productos FarmaceuticosDocument9 pagesAdquisicion de Los Productos FarmaceuticosMelina Elgegren zavalagaNo ratings yet
- Aparato Reproductor MasculinoDocument30 pagesAparato Reproductor MasculinoGLADYSNo ratings yet
- Informe Constantes FisilogicasDocument6 pagesInforme Constantes FisilogicasAlex UribeNo ratings yet
- Capítulo 36.Document40 pagesCapítulo 36.Leonel Gonthier100% (1)
- TecnovigilanciaDMDocument26 pagesTecnovigilanciaDMRicardo Robles100% (1)
- Clase - 2 y 3 PDFDocument79 pagesClase - 2 y 3 PDFMiguel Rodas Salazar100% (1)
- Análisis Del Mercado Farmacéutico en El EcuadorDocument2 pagesAnálisis Del Mercado Farmacéutico en El EcuadorEmily DeyaneiraNo ratings yet
- Conceptos Legales de Productos FarmaceuticosDocument10 pagesConceptos Legales de Productos FarmaceuticosVictor Hugo Soto ArriagadaNo ratings yet
- Implementacion de La Dosis Unitaria-Equipo UrmDocument21 pagesImplementacion de La Dosis Unitaria-Equipo UrmGian Santa CruzNo ratings yet
- Los GlucidosDocument22 pagesLos GlucidosAnonymous 5P0Wkmiu9eNo ratings yet
- CLASE 6 - Evaluación Del Costo de Los Medicamentos 2018Document44 pagesCLASE 6 - Evaluación Del Costo de Los Medicamentos 2018diana vidalNo ratings yet
- 3 Productos de Venta Libre Otc y Productos NaturalesDocument7 pages3 Productos de Venta Libre Otc y Productos NaturalesJoselyn VillanoNo ratings yet
- ENVENENAMIENTODocument11 pagesENVENENAMIENTOGianella Echevarría100% (1)
- Legislacion y Deontología FarmaceuticaDocument17 pagesLegislacion y Deontología FarmaceuticaAlejandra Gutierrez ʚïɞNo ratings yet
- Presentacion Proyecto VENCIDOS CRS11Document11 pagesPresentacion Proyecto VENCIDOS CRS11diomedesblancoNo ratings yet
- Manual de Buenas Practicas de Manufarctura Grupo FsDocument21 pagesManual de Buenas Practicas de Manufarctura Grupo FsGene Flores CauperNo ratings yet
- BPDDocument20 pagesBPDHernan Velez RiosNo ratings yet
- Atb Parteras PDFDocument24 pagesAtb Parteras PDFAna Maria PedrogaNo ratings yet
- Manual BPM en Galénicos (Tesis)Document33 pagesManual BPM en Galénicos (Tesis)Edwin FloresNo ratings yet
- AnticonceptivosDocument46 pagesAnticonceptivosblueNo ratings yet
- Clase 14DM - TECNOVIGILANCIA.Document27 pagesClase 14DM - TECNOVIGILANCIA.FIORELLANo ratings yet
- Funcionamiento y Control BotiquinesDocument38 pagesFuncionamiento y Control BotiquinesBernat Bertomeu BarralNo ratings yet
- Eett SieDocument23 pagesEett SieLobojNo ratings yet
- Normas farmacias y boticasDocument25 pagesNormas farmacias y boticassismed2No ratings yet
- Materiales InflamablesDocument13 pagesMateriales InflamablesCristhian Daniel Suarez ZamoraNo ratings yet
- Terminología FarmacéuticaDocument22 pagesTerminología FarmacéuticaMELANY GIRALDO SAENZNo ratings yet
- Grupo 1. Eficacia, Efectividad y Falla Terapéutica - KeoDocument8 pagesGrupo 1. Eficacia, Efectividad y Falla Terapéutica - KeoAnatoweb0% (1)
- Cuestionario 5 To ModuloDocument7 pagesCuestionario 5 To ModuloCristhian JimenezNo ratings yet
- Acta de Inspeccion Botica y FarmaciaDocument10 pagesActa de Inspeccion Botica y FarmaciaLUIS ANDRES DURAND BELENNo ratings yet
- Características de prescripción de antibióticos en recetas médicas del HNDMDocument96 pagesCaracterísticas de prescripción de antibióticos en recetas médicas del HNDMPedro Pablo TorresNo ratings yet
- 2.3.-Reglamento de EF DS 014-2011 DS 02-2012 y DS 033-2014Document28 pages2.3.-Reglamento de EF DS 014-2011 DS 02-2012 y DS 033-2014Deyser Hernan Fernandez CenturionNo ratings yet
- Practican3 Anlisiscualitativoycuantitativodelacefalexina 151121022721 Lva1 App6891Document8 pagesPractican3 Anlisiscualitativoycuantitativodelacefalexina 151121022721 Lva1 App6891Yuvil ReateguiNo ratings yet
- Dr. Cesar Amaro - Manual BpofDocument41 pagesDr. Cesar Amaro - Manual BpofFranklyn Torres Chacon100% (1)
- 3103 - Venta y Dispensacion Prod. NaturalesDocument35 pages3103 - Venta y Dispensacion Prod. NaturalesGiancarlo YoveraNo ratings yet
- Dispositivos MédicosDocument12 pagesDispositivos MédicosLenin RiveraNo ratings yet
- Formas Farmaceuticas SolidasDocument23 pagesFormas Farmaceuticas SolidasNAYELLY LAUREANO FASABINo ratings yet
- Normas para La Apertura de Una Oficina FarmaceuticaDocument12 pagesNormas para La Apertura de Una Oficina FarmaceuticaJuan Buendia100% (1)
- Poes 12Document3 pagesPoes 12Mariela Livia MoralesNo ratings yet
- AINESDocument41 pagesAINESSergio David Vargas Choqueticlla100% (1)
- Factores Que Influyen en La Composición Química de Productos FitoterapéuticosDocument5 pagesFactores Que Influyen en La Composición Química de Productos FitoterapéuticosDavi RojasNo ratings yet
- Capacitacion No.1 Dependiente de Farmacia Feb2014Document14 pagesCapacitacion No.1 Dependiente de Farmacia Feb2014Marvin ZacariasNo ratings yet
- Reconocimiento de Medicamentos IlegalesDocument18 pagesReconocimiento de Medicamentos IlegalesRene ZevallosNo ratings yet
- Clase 1 BiofarmaciaDocument28 pagesClase 1 BiofarmaciaIván CastroNo ratings yet
- TEMA 18 APLICACIONES DE LOS PRODUCTOS NATURALES Parte 1Document48 pagesTEMA 18 APLICACIONES DE LOS PRODUCTOS NATURALES Parte 1elmeras1987No ratings yet
- Sistema Peruano de FarmacovigilanciaDocument44 pagesSistema Peruano de FarmacovigilanciaDiego Ale MauricioNo ratings yet
- Calendula OfficinalisDocument4 pagesCalendula OfficinalisLia Gabriela GonzálezNo ratings yet
- Sistemas de dispensación hospitalariaDocument54 pagesSistemas de dispensación hospitalariaFrancisco ReyesNo ratings yet
- Cuidados pacientes trastornos hematológicos oncológicosDocument30 pagesCuidados pacientes trastornos hematológicos oncológicosadriana lermaNo ratings yet
- Ley de PsicotropicosDocument25 pagesLey de PsicotropicosCESMO ESTELINo ratings yet
- Manual de Buenas Practicas de Almacenamiento de Insumos MedicosDocument27 pagesManual de Buenas Practicas de Almacenamiento de Insumos MedicosHugo Ulloa DiazNo ratings yet
- Manual de BPM de Productos Galenicos y Recursos Terapeuticos Naturales-ListoDocument25 pagesManual de BPM de Productos Galenicos y Recursos Terapeuticos Naturales-Listoluis alberto valera camposNo ratings yet
- Buenas Practicas de Produccion (Leer)Document3 pagesBuenas Practicas de Produccion (Leer)julia camara manuelNo ratings yet
- Modelo DipaosDocument3 pagesModelo DipaosAlonso SardónNo ratings yet
- Diapos BPMDocument10 pagesDiapos BPMAlonso SardónNo ratings yet
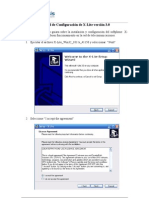
- Manual de Configuración de X-LiteDocument8 pagesManual de Configuración de X-LiteAlonso SardónNo ratings yet
- Acupuntura Ying-Yang (4 Pag) PDFDocument4 pagesAcupuntura Ying-Yang (4 Pag) PDFAlejandra Hurtado CuevasNo ratings yet
- Acupuntura Cinco Movimientos (4 Pag)Document4 pagesAcupuntura Cinco Movimientos (4 Pag)titosol100% (1)
- 234 Bioquimik SabeeeDocument22 pages234 Bioquimik SabeeeAlonso SardónNo ratings yet
- Receptores de MembranaDocument17 pagesReceptores de MembranaUriel YépezNo ratings yet
- Pintura sobre sal: aprende una nueva técnica artísticaDocument2 pagesPintura sobre sal: aprende una nueva técnica artísticaAUGUSTONo ratings yet
- Exp MatrizDocument4 pagesExp MatrizPepe LaporteNo ratings yet
- Leonel Ortiz Mauricio Act2Document7 pagesLeonel Ortiz Mauricio Act2Leonel Ortiz MauricioNo ratings yet
- Opcion Acaso 06Document7 pagesOpcion Acaso 06andresoihfhhs50% (2)
- Organización Del Sistema Nervioso, Neurona-Sinapsis PDFDocument63 pagesOrganización Del Sistema Nervioso, Neurona-Sinapsis PDFPerla LPNo ratings yet
- Mi Ángel C2 2024-1Document192 pagesMi Ángel C2 2024-1ordonezmagdalena75No ratings yet
- Academia Del Software Libre en El Marco de Una Politica Institucional de Desarrollo Regional de Las Tecnologias de InformaciónDocument7 pagesAcademia Del Software Libre en El Marco de Una Politica Institucional de Desarrollo Regional de Las Tecnologias de InformaciónYudannys PoloNo ratings yet
- Instituto Ecuatoriano de Seguridad Social ClaveDocument2 pagesInstituto Ecuatoriano de Seguridad Social ClaveBISMARNo ratings yet
- Examen Matemáticas III 2da VueltaDocument3 pagesExamen Matemáticas III 2da VueltaWenzel Dietrich ValentinaNo ratings yet
- Casos Prácticos de Contabilidad de FranquiciaDocument2 pagesCasos Prácticos de Contabilidad de FranquiciaLuis Alfonso Magaña ramosNo ratings yet
- Diagnóstico ambiental comunitario: Herramientas para su ejecuciónDocument3 pagesDiagnóstico ambiental comunitario: Herramientas para su ejecuciónRosario Orrego de BaldizonNo ratings yet
- Planificación del proceso deportivo en escuelas mediante programas de enseñanzaDocument5 pagesPlanificación del proceso deportivo en escuelas mediante programas de enseñanzaAlejandro Romero CruzNo ratings yet
- 1 y 2. Concepto e Import An CIA Del SueloDocument55 pages1 y 2. Concepto e Import An CIA Del Sueloands_120No ratings yet
- Listado de Inmuebles en La Zona Central de San Salvador IIDocument52 pagesListado de Inmuebles en La Zona Central de San Salvador IIKrla DunkelsternNo ratings yet
- Color AntesDocument13 pagesColor AntesRuby TuesdayNo ratings yet
- Maduración de La AfinidadDocument2 pagesMaduración de La AfinidadMarieta MOreira PincayNo ratings yet
- Ascenso Capilar en CuñasDocument2 pagesAscenso Capilar en CuñasLr FrNo ratings yet
- Implementación de programación paralela con MPIDocument9 pagesImplementación de programación paralela con MPIOrlando Alcala Vargas100% (1)
- Letras de Los Himnos CostarricensesDocument12 pagesLetras de Los Himnos CostarricensesJorge SotoNo ratings yet
- Formato PP2 09Document4 pagesFormato PP2 09Guerra Kay TanNo ratings yet
- Matrícula TECSUP 2022-2Document13 pagesMatrícula TECSUP 2022-2santiagoNo ratings yet
- Taller Fundamentos ElectricosDocument4 pagesTaller Fundamentos Electricosjose estebanNo ratings yet
- Quiz 2 Gerencia Financiera Semana 7Document7 pagesQuiz 2 Gerencia Financiera Semana 7lina rubianoNo ratings yet
- Omar Leon - Gerardo Toscano - Trabajo de Investigacion - Bachiller - 2020Document333 pagesOmar Leon - Gerardo Toscano - Trabajo de Investigacion - Bachiller - 2020Brady19No ratings yet
- 11 BenchmarkingDocument28 pages11 Benchmarkingoscar caballeroNo ratings yet
- Solución EBAU CyL Física 2021 Junio V2Document6 pagesSolución EBAU CyL Física 2021 Junio V2sweetkairi1992No ratings yet