You might also like
- SGB Amravati University B.Pharm Pharmacovigilance Question BankDocument10 pagesSGB Amravati University B.Pharm Pharmacovigilance Question BankTabassum PopatpotraNo ratings yet
- Historical Perspective, Different Phases of Clinical TrialsDocument16 pagesHistorical Perspective, Different Phases of Clinical Trialsexcel proNo ratings yet
- MCQ's - Clinical ResearchDocument4 pagesMCQ's - Clinical Researchdivya83% (12)
- Differences between Indian GCP and ICH-GCP guidelinesDocument1 pageDifferences between Indian GCP and ICH-GCP guidelinesankit geete50% (2)
- ICH GCP & Indian Clinical Trial GuidelineDocument97 pagesICH GCP & Indian Clinical Trial GuidelineRanjeet PrasadNo ratings yet
- Question PapersDocument5 pagesQuestion PapersBiswajeet DasguptaNo ratings yet
- Drug Safety Associate Job Interview Questions and AnswersDocument8 pagesDrug Safety Associate Job Interview Questions and AnswersGaurav Kumar100% (1)
- Difference Between Indian GCP and ICHGCPDocument5 pagesDifference Between Indian GCP and ICHGCPsuri333No ratings yet
- 3 Who Drug DictionaryDocument42 pages3 Who Drug DictionaryTesfa TesfaNo ratings yet
- Clinicaltrials Presentation 111011025509 Phpapp02 PDFDocument51 pagesClinicaltrials Presentation 111011025509 Phpapp02 PDFGayathri Parthasarathy100% (1)
- Clinical Trial DesignsDocument18 pagesClinical Trial DesignsVenkatesh Gavini100% (1)
- Assignment 3: Literature Review: Unit 1 - Basic Course in Biomedical Research: Cycle 2 (Mar-Jun 2020)Document6 pagesAssignment 3: Literature Review: Unit 1 - Basic Course in Biomedical Research: Cycle 2 (Mar-Jun 2020)stNo ratings yet
- RM SyllabusDocument4 pagesRM SyllabuskoduruNo ratings yet
- Assignment 2: Formulating Research: Unit 1 - Basic Course in Biomedical Research: Cycle 2 (Mar-Jun 2020)Document6 pagesAssignment 2: Formulating Research: Unit 1 - Basic Course in Biomedical Research: Cycle 2 (Mar-Jun 2020)stNo ratings yet
- Ich-Gcp & Schedule yDocument38 pagesIch-Gcp & Schedule ySahiti PendyalaNo ratings yet
- International Regulatory Requirements On Clinical Trails and Data ManagementDocument19 pagesInternational Regulatory Requirements On Clinical Trails and Data ManagementJay PraveenNo ratings yet
- Clinical TrialsDocument20 pagesClinical Trialsvamsykrishnabj0% (1)
- Quality indicators improve anesthesia careDocument5 pagesQuality indicators improve anesthesia caredrskumarNo ratings yet
- GSK b2c109575 Clinical Study Report Redact PDFDocument1,524 pagesGSK b2c109575 Clinical Study Report Redact PDFdhananjayNo ratings yet
- Ich Guidelines: #Fouziya's-NotesDocument3 pagesIch Guidelines: #Fouziya's-NotesFouziya BegumNo ratings yet
- Feulgen Stain QuestionsDocument5 pagesFeulgen Stain QuestionsFatma AdelNo ratings yet
- Anti Urolithiatic Activity of Cucumis Fruit ExtractDocument7 pagesAnti Urolithiatic Activity of Cucumis Fruit ExtractRajkiran EllandalaNo ratings yet
- ICH-GCP guidelines and clinical researchDocument58 pagesICH-GCP guidelines and clinical researchAkhil SoodNo ratings yet
- 06 - Chapter 1Document37 pages06 - Chapter 1Vikash KushwahaNo ratings yet
- Internal DiseaseDocument55 pagesInternal Diseaseshilpa sekhar278No ratings yet
- Multiple Choice Questions On Research Methodology - MCQ Biology - Learning Biology Through MCQsDocument4 pagesMultiple Choice Questions On Research Methodology - MCQ Biology - Learning Biology Through MCQspink1231No ratings yet
- MRU EXAM 2015 MASTER ONE UNIV MSILA BERBACHE SDocument8 pagesMRU EXAM 2015 MASTER ONE UNIV MSILA BERBACHE Sbersam05No ratings yet
- Chapter 18 - Self-Assessment Q&ADocument6 pagesChapter 18 - Self-Assessment Q&AGustavo BobadillaNo ratings yet
- Family Medicine LogbookDocument2 pagesFamily Medicine LogbookDr Onn Azli PuadeNo ratings yet
- Assignment 4: Measures of Disease Frequency: Unit 1 - Basic Course in Biomedical Research: Cycle 2 (Mar-Jun 2020)Document6 pagesAssignment 4: Measures of Disease Frequency: Unit 1 - Basic Course in Biomedical Research: Cycle 2 (Mar-Jun 2020)stNo ratings yet
- Pharmacology III Exam QuestionsDocument4 pagesPharmacology III Exam QuestionsTanvi MalewarNo ratings yet
- Validity of Epidemiological StudiesDocument6 pagesValidity of Epidemiological StudiesstNo ratings yet
- Health Research FuundamentalsDocument57 pagesHealth Research FuundamentalsNavitha N100% (2)
- Toxicology Q-Bank Updated VersionDocument18 pagesToxicology Q-Bank Updated VersionAhmed Mohamed100% (1)
- Study DesignDocument45 pagesStudy DesignDeepak SharmaNo ratings yet
- Clinical Research Study Questions and AnswersDocument3 pagesClinical Research Study Questions and AnswersPartha100% (2)
- Schedule YDocument11 pagesSchedule YRamling Patrakar100% (1)
- Teaching Plan HCPDocument5 pagesTeaching Plan HCPNaveen ChoudharyNo ratings yet
- GCP protects human subjects in clinical researchDocument3 pagesGCP protects human subjects in clinical researchSok YeeNo ratings yet
- Niper NotesDocument9 pagesNiper NotesVizit DubeyNo ratings yet
- Importance of Pharmacovigilance For Pharmaceutical IndustryDocument24 pagesImportance of Pharmacovigilance For Pharmaceutical IndustryPiratesNo ratings yet
- Clinical Trial PhasesDocument4 pagesClinical Trial PhasesManish SarasvatiNo ratings yet
- Lesson - Types of StudiesDocument34 pagesLesson - Types of StudiesVikashgtmNo ratings yet
- Biostatistics and Research MethodsDocument2 pagesBiostatistics and Research MethodsOsama JavedNo ratings yet
- Basic Pharmacovigilance Interview Questions For FRESHERSDocument8 pagesBasic Pharmacovigilance Interview Questions For FRESHERSPrakash MishraNo ratings yet
- 10 - Drug DevelopmentDocument5 pages10 - Drug DevelopmentLaura SaglietiNo ratings yet
- EthicsDocument65 pagesEthicsUmesh KawalkarNo ratings yet
- Fda Ind FormDocument3 pagesFda Ind Formapi-278781792100% (1)
- Success & Excellence: in Clinical ResearchDocument4 pagesSuccess & Excellence: in Clinical ResearchNarayana NaveenkumarNo ratings yet
- Qualitative Research Methods - An OverviewDocument6 pagesQualitative Research Methods - An OverviewstNo ratings yet
- Oxford University Press - Online Resource Centre - Multiple Choice QuestionsDocument3 pagesOxford University Press - Online Resource Centre - Multiple Choice QuestionsMuhammad haseebNo ratings yet
- Validating The Link Between Aetiology, Pathology, Diagnosis, and Treatmentin Relation To The Philosophical Principles of TibbDocument12 pagesValidating The Link Between Aetiology, Pathology, Diagnosis, and Treatmentin Relation To The Philosophical Principles of TibbIJAR JOURNALNo ratings yet
- Recommended Format For A Research ProtocolDocument2 pagesRecommended Format For A Research ProtocolLea EdwardsNo ratings yet
- Ÿßÿ ŸäŸîŸÑŸá ŸÖŸáŸÖŸá Study DesignDocument3 pagesŸßÿ ŸäŸîŸÑŸá ŸÖŸáŸÖŸá Study DesignaliNo ratings yet
- Sumy State University Lecture on AllergiesDocument56 pagesSumy State University Lecture on AllergiesTazrian FarahNo ratings yet
- Clinical Trial DocsDocument44 pagesClinical Trial DocspalanivelNo ratings yet
- B Pharm 3rd SemDocument14 pagesB Pharm 3rd SemKoushik PaulNo ratings yet
- Interview Questions For PharmacovigilanceDocument11 pagesInterview Questions For PharmacovigilanceRumaisa Wasi100% (2)
- Notification UPSC Advt No 04 by 2015 Drugs Inspector Asst Professor and Other PostsDocument10 pagesNotification UPSC Advt No 04 by 2015 Drugs Inspector Asst Professor and Other PostsMinuChakrabortyNo ratings yet
- Book 3457Document5 pagesBook 3457Umesh ChikhlikarNo ratings yet
- Book 1345Document5 pagesBook 1345Umesh ChikhlikarNo ratings yet
- Book 123456Document5 pagesBook 123456Umesh ChikhlikarNo ratings yet
- General KnowledgeDocument4 pagesGeneral KnowledgeUmesh ChikhlikarNo ratings yet
- Book1 WeightDocument5 pagesBook1 WeightUmesh ChikhlikarNo ratings yet
- Spectrum &combDocument1 pageSpectrum &combUmesh ChikhlikarNo ratings yet
- Load CombinationDocument2 pagesLoad CombinationUmesh ChikhlikarNo ratings yet
- International Codes-Is456 - Beam Design in STAAD Pro - V8iDocument5 pagesInternational Codes-Is456 - Beam Design in STAAD Pro - V8iUmesh ChikhlikarNo ratings yet
- Area of ShapesDocument9 pagesArea of ShapesUmesh Chikhlikar100% (1)
- Column Design per IS 13920Document11 pagesColumn Design per IS 13920Umesh ChikhlikarNo ratings yet
- Book2 ReactionsDocument1 pageBook2 ReactionsUmesh ChikhlikarNo ratings yet
- General KnowledgeDocument4 pagesGeneral KnowledgeUmesh ChikhlikarNo ratings yet
- NCDEX commodity futures prices and margin detailsDocument6 pagesNCDEX commodity futures prices and margin detailsUmesh ChikhlikarNo ratings yet
- AreaDocument1 pageAreaUmesh ChikhlikarNo ratings yet
- StructstdsDocument44 pagesStructstdsMuraleedharanNo ratings yet
- MCX SpanDocument6 pagesMCX SpanUmesh ChikhlikarNo ratings yet
- General Aptitude: 1. Chile 2. Russia 3. South Africa 4. China Ans: 1Document26 pagesGeneral Aptitude: 1. Chile 2. Russia 3. South Africa 4. China Ans: 1Prashanth SbNo ratings yet
- Sugar Fermentation TestDocument15 pagesSugar Fermentation TestUmesh Chikhlikar67% (3)
- NCDEX commodity futures prices and margin detailsDocument6 pagesNCDEX commodity futures prices and margin detailsUmesh ChikhlikarNo ratings yet
- MCX SpanDocument6 pagesMCX SpanUmesh ChikhlikarNo ratings yet
- LPR ProjDocument6 pagesLPR ProjUmesh ChikhlikarNo ratings yet
- Stability CertificateDocument1 pageStability CertificateUmesh ChikhlikarNo ratings yet
- Microbiology TestingDocument15 pagesMicrobiology TestingUmesh ChikhlikarNo ratings yet
- MCX SpanDocument6 pagesMCX SpanUmesh ChikhlikarNo ratings yet
- Ebook 2013Document30 pagesEbook 2013Umesh ChikhlikarNo ratings yet
- MCX SpanDocument6 pagesMCX SpanUmesh ChikhlikarNo ratings yet
- LPR ProjDocument6 pagesLPR ProjUmesh ChikhlikarNo ratings yet
- MCX SpanDocument6 pagesMCX SpanUmesh ChikhlikarNo ratings yet
- MCX SpanDocument6 pagesMCX SpanUmesh ChikhlikarNo ratings yet
- Sample GCP ChecklistDocument8 pagesSample GCP Checklistsreeraj.guruvayoor100% (1)
- Digital vaccine certificate detailsDocument1 pageDigital vaccine certificate detailsJoeanna JoeannaNo ratings yet
- NEET 2020 Provisional Result List For Round 2 of Under Graduate Courses PDFDocument730 pagesNEET 2020 Provisional Result List For Round 2 of Under Graduate Courses PDFDollyNo ratings yet
- Weekly Sales Performance and Product Promotion ReportDocument6 pagesWeekly Sales Performance and Product Promotion ReportZelalemNo ratings yet
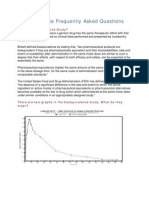
- Bioequivalence Frequently Asked Questions: What Is Bioequivalence Study?Document4 pagesBioequivalence Frequently Asked Questions: What Is Bioequivalence Study?Ahmed HasanNo ratings yet
- CUI Texto Examen Multimedia Nivel 6 Curso BasicoDocument2 pagesCUI Texto Examen Multimedia Nivel 6 Curso BasicobertoniramiroNo ratings yet
- Jourding SIRS and pSOFADocument3 pagesJourding SIRS and pSOFASUARDI SARIBUNo ratings yet
- Use of Placebo Controls in Clinical Research - Trials2012Document7 pagesUse of Placebo Controls in Clinical Research - Trials2012Ju ChangNo ratings yet
- 1 - Introduction To Drug Development For 2022 Lecture1Document14 pages1 - Introduction To Drug Development For 2022 Lecture1addisu afrassaNo ratings yet
- Clinical Trials PDFDocument17 pagesClinical Trials PDFBin Hip100% (2)
- Drug DevelopmentDocument26 pagesDrug DevelopmentshraddhaJPNo ratings yet
- Drug Development ProcessDocument65 pagesDrug Development ProcessMonika AhlavadiNo ratings yet
- Prescriptions Labelling Latin Terms FA 20120222Document24 pagesPrescriptions Labelling Latin Terms FA 20120222SherazButtNo ratings yet
- PENGANTAR TAHAPAN PEMETAAN SISTEMATIS: Bibliometrics Studies, Systematic Mapping Studies, Systematic Literature Review, Dan Scoping Review StudiesDocument7 pagesPENGANTAR TAHAPAN PEMETAAN SISTEMATIS: Bibliometrics Studies, Systematic Mapping Studies, Systematic Literature Review, Dan Scoping Review StudiesAhmad BaiquniNo ratings yet
- RD Brochure 022307Document14 pagesRD Brochure 022307Kevyn W WangNo ratings yet
- PharmaSUG XXDocument12 pagesPharmaSUG XXhhjbjhbNo ratings yet
- Overview of Randomized Controlled Trials: Review ArticleDocument7 pagesOverview of Randomized Controlled Trials: Review ArticleCut Sinta BeliaNo ratings yet
- C - II - 3 - Critical AppraisalDocument10 pagesC - II - 3 - Critical AppraisalWahyu NandaNo ratings yet
- Standard Operating Procedure For The Recording, Management and Reporting of Adverse Events by InvestigatorsDocument19 pagesStandard Operating Procedure For The Recording, Management and Reporting of Adverse Events by Investigatorsvikram kushwahaNo ratings yet
- CR IntroDocument21 pagesCR Introsivaram_12213443No ratings yet
- Clinical Study Report Template - GHTDocument22 pagesClinical Study Report Template - GHThpradeep100% (1)
- Clinical Trial Safety MonitoringDocument3 pagesClinical Trial Safety MonitoringShivananda PradhanNo ratings yet
- Introduction and Evaluation of Pharmacovigilance For BeginnersDocument8 pagesIntroduction and Evaluation of Pharmacovigilance For BeginnersShaun MerchantNo ratings yet
- Drug DPMTDocument27 pagesDrug DPMTapi-3842711100% (1)
- High-quality evidence source for healthcare decisionsDocument19 pagesHigh-quality evidence source for healthcare decisionsBrankaPodgoricaNo ratings yet
- Pharmacovigilance: A ReviewDocument4 pagesPharmacovigilance: A ReviewMahewash PathanNo ratings yet
- DosisaplicadasDocument55 pagesDosisaplicadasLuis Alfredo Cansimance LopezNo ratings yet
- Clinical Trials in India: A Critical EvaluationDocument10 pagesClinical Trials in India: A Critical EvaluationRakesh K SNo ratings yet
- Requirements: For A Valid Vaccination Certificate (3 Doses)Document1 pageRequirements: For A Valid Vaccination Certificate (3 Doses)HaoYang ChiaNo ratings yet
- The Four Phases of Clinical Trials - June 2016 1Document1 pageThe Four Phases of Clinical Trials - June 2016 1Mihir OzaNo ratings yet