You might also like
- Synthesis and Characterization of PEG Graft Quaternized Chitosan and Cationic Polymeric Liposomes For Drug Delivery Liang XDocument8 pagesSynthesis and Characterization of PEG Graft Quaternized Chitosan and Cationic Polymeric Liposomes For Drug Delivery Liang Xalchemik1515No ratings yet
- Synthesis, Characterization and Cytotoxicity of Poly (Ethylene Glycol) - Graft-Trimethyl Chitosan Block CopolymersDocument14 pagesSynthesis, Characterization and Cytotoxicity of Poly (Ethylene Glycol) - Graft-Trimethyl Chitosan Block Copolymersalchemik1515No ratings yet
- Synthesis and Characterization of Graft Copolymer of Chitosan and Polyethylene Glycol Zhao YDocument8 pagesSynthesis and Characterization of Graft Copolymer of Chitosan and Polyethylene Glycol Zhao Yalchemik1515No ratings yet
- Synthesis and Charn of in Situ Cross-Linked Hydrogel Based On Self-Assembly of Thiol-Modified Chitosan With PEG DiacrylateDocument8 pagesSynthesis and Charn of in Situ Cross-Linked Hydrogel Based On Self-Assembly of Thiol-Modified Chitosan With PEG Diacrylatealchemik1515No ratings yet
- Synthesis of Cross-Linked N - (2-Carboxybenzyl) Chitosan PH Sensitive PolyelectrolyteDocument8 pagesSynthesis of Cross-Linked N - (2-Carboxybenzyl) Chitosan PH Sensitive Polyelectrolytealchemik1515No ratings yet
- Synthesis, Characterization, and Cytotoxicity of TMC-graft-poly (Vinyl Alcohol) CopolymersDocument8 pagesSynthesis, Characterization, and Cytotoxicity of TMC-graft-poly (Vinyl Alcohol) Copolymersalchemik1515No ratings yet
- Synthesis of Zirconium Carbide Powders Using Chitosan As Carbon SourceDocument4 pagesSynthesis of Zirconium Carbide Powders Using Chitosan As Carbon Sourcealchemik1515No ratings yet
- Synthesis and Characterization of Poly (Amino Urea Urethane) - Based Block Copolymer and Its Potential Application As Injectable PH-temperature-sensitive Hydrogel For Protein CarrierDocument7 pagesSynthesis and Characterization of Poly (Amino Urea Urethane) - Based Block Copolymer and Its Potential Application As Injectable PH-temperature-sensitive Hydrogel For Protein Carrieralchemik1515No ratings yet
- Supercritical Fluid Technologies and Tissue Engineering ScaffoldsDocument9 pagesSupercritical Fluid Technologies and Tissue Engineering Scaffoldsalchemik1515No ratings yet
- Synthesis and Characterization of A Novel Amphiphilic Chitosan-Polylactide Graft CopolymerDocument7 pagesSynthesis and Characterization of A Novel Amphiphilic Chitosan-Polylactide Graft Copolymeralchemik1515No ratings yet
- Synthesis and Characterization of Temp and PH - Sensitive Hydrogels Based On Chitosan-Grafted NIPAAm RadiationDocument5 pagesSynthesis and Characterization of Temp and PH - Sensitive Hydrogels Based On Chitosan-Grafted NIPAAm Radiationalchemik1515No ratings yet
- Swelling Behavior of Polyacrylamide Laponite Clay Nanocomposite Hydrogels PH Sensitive Property 2009Document9 pagesSwelling Behavior of Polyacrylamide Laponite Clay Nanocomposite Hydrogels PH Sensitive Property 2009alchemik1515No ratings yet
- Synthesis and Characteristics of Biodegradable and Temperature Responsive Polymeric Micelles Based On Poly (Aspartic Acid) - G-Poly (N-Isopropylacrylamide-co-N, N-Dimethylacrylamide)Document32 pagesSynthesis and Characteristics of Biodegradable and Temperature Responsive Polymeric Micelles Based On Poly (Aspartic Acid) - G-Poly (N-Isopropylacrylamide-co-N, N-Dimethylacrylamide)alchemik1515No ratings yet
- Synthesis and Characterization of Folic Acid Modified Water-Soluble Chitosan Derivatives For Folate-Receptor-Mediated TargetingDocument7 pagesSynthesis and Characterization of Folic Acid Modified Water-Soluble Chitosan Derivatives For Folate-Receptor-Mediated Targetingalchemik1515No ratings yet
- Synthesis and Characterisation of Polyacrylamide-Laponite Nanocomposite Hydrogels2Document7 pagesSynthesis and Characterisation of Polyacrylamide-Laponite Nanocomposite Hydrogels2alchemik1515No ratings yet
- Preparation and Characterization of Poly (Ethylene Glycol) G Chitosan With Water and Organosolubility Hu YDocument8 pagesPreparation and Characterization of Poly (Ethylene Glycol) G Chitosan With Water and Organosolubility Hu Yalchemik1515No ratings yet
- Synthesis and Characterisation of Polyacrylamide-Laponite Nanocomposite HydrogelsDocument36 pagesSynthesis and Characterisation of Polyacrylamide-Laponite Nanocomposite Hydrogelsalchemik1515No ratings yet
- Stimuli-Responsive Microgels For The Loading and Release of Functional Compounds - Fundamental Concepts and ApplicationsDocument23 pagesStimuli-Responsive Microgels For The Loading and Release of Functional Compounds - Fundamental Concepts and Applicationsalchemik1515No ratings yet
- Preparation and Characterization of Folate-Poly (Ethylene Glycol) - Grafted-Trimethylchitosan For Intracellular Transport of Protein Through Folate Receptor-Mediated EndocytosisDocument7 pagesPreparation and Characterization of Folate-Poly (Ethylene Glycol) - Grafted-Trimethylchitosan For Intracellular Transport of Protein Through Folate Receptor-Mediated Endocytosisalchemik1515No ratings yet
- Recent Progress On Study of Hybrid Hydrogels For Water TreatmentDocument9 pagesRecent Progress On Study of Hybrid Hydrogels For Water Treatmentalchemik1515No ratings yet
- Rapid Synthesis and Characterization of Chitosan G Poly (D, L Lactide) Copolymers With Hydroxyethyl Chitosan As A Macroinitiator Under Microwave IrradiationDocument7 pagesRapid Synthesis and Characterization of Chitosan G Poly (D, L Lactide) Copolymers With Hydroxyethyl Chitosan As A Macroinitiator Under Microwave Irradiationalchemik1515No ratings yet
- PH Sensitive Nanoparticles Self Assembled From A Novel Class of Biodegradable Amphiphilic Copolymers Based On Chitosan Cai GDocument6 pagesPH Sensitive Nanoparticles Self Assembled From A Novel Class of Biodegradable Amphiphilic Copolymers Based On Chitosan Cai Galchemik1515No ratings yet
- Preparation and Characterization of Water-Soluble Chitin and Chitosan DerivativesDocument11 pagesPreparation and Characterization of Water-Soluble Chitin and Chitosan Derivativesalchemik1515No ratings yet
- Rapid Synthesis and Characterization of Chitosan G Poly (D, L Lactide) Copolymers With Hydroxyethyl Chitosan As A Macroinitiator Under Microwave IrradiationDocument7 pagesRapid Synthesis and Characterization of Chitosan G Poly (D, L Lactide) Copolymers With Hydroxyethyl Chitosan As A Macroinitiator Under Microwave Irradiationalchemik1515No ratings yet
- Preparation and Characterization of Novel Hybrid of Chitosan G Lactic Acid and Montmorillonite 2006 Journal of Biomedical Materials Research Part ADocument11 pagesPreparation and Characterization of Novel Hybrid of Chitosan G Lactic Acid and Montmorillonite 2006 Journal of Biomedical Materials Research Part Aalchemik1515No ratings yet
- Preparation and Optimization of PMAA-Chitosan-PEG Nanoparticles For Oral Drug DeliveryDocument7 pagesPreparation and Optimization of PMAA-Chitosan-PEG Nanoparticles For Oral Drug Deliveryalchemik1515No ratings yet
- Preparation and Characterization of Poly (Ethylene Glycol) G Chitosan With Water and Organosolubility Hu YDocument8 pagesPreparation and Characterization of Poly (Ethylene Glycol) G Chitosan With Water and Organosolubility Hu Yalchemik1515No ratings yet
- PH Sensitive Genipin Cross Linked Chitosan Microspheres For Heparin Removal 2008 BiomacromoleculesDocument6 pagesPH Sensitive Genipin Cross Linked Chitosan Microspheres For Heparin Removal 2008 Biomacromoleculesalchemik1515No ratings yet
- Poly (Ethylene Glycol) - Carboxymethyl Chitosan-Based PH-responsive Hydrogels - Photo-Induced Synthesis, Characterization, Swelling, and in Vitro Evaluation As Potential Drug CarriersDocument9 pagesPoly (Ethylene Glycol) - Carboxymethyl Chitosan-Based PH-responsive Hydrogels - Photo-Induced Synthesis, Characterization, Swelling, and in Vitro Evaluation As Potential Drug Carriersalchemik1515No ratings yet
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Cost of FasdDocument12 pagesCost of FasdMary Harber-IlesNo ratings yet
- BIOL 4430 Syllabus Curr Bauzon S13v1Document5 pagesBIOL 4430 Syllabus Curr Bauzon S13v1Angela ReyesNo ratings yet
- LESSON PLAN EnglishDocument7 pagesLESSON PLAN EnglishMarie Antonette Aco BarbaNo ratings yet
- Star Fleet Ships of The Four Years War, Volume 2Document28 pagesStar Fleet Ships of The Four Years War, Volume 2Rob RobertsNo ratings yet
- Halo: The Empyrean EffectDocument6 pagesHalo: The Empyrean EffectStephen PerezNo ratings yet
- Judith Butler: Performative AgencyDocument16 pagesJudith Butler: Performative Agencyjacquesfatalist100% (2)
- The Learner Understands The Guidelines and Criteria in The Selection and Evaluation of Health InformationDocument3 pagesThe Learner Understands The Guidelines and Criteria in The Selection and Evaluation of Health InformationANACORITA O. SILAGANNo ratings yet
- Higher Unit 06b Check in Test Linear Graphs Coordinate GeometryDocument7 pagesHigher Unit 06b Check in Test Linear Graphs Coordinate GeometryPrishaa DharnidharkaNo ratings yet
- 16. SO2ndEdAAchTest5.doc - Google ДокументиDocument4 pages16. SO2ndEdAAchTest5.doc - Google ДокументиAlinaNo ratings yet
- Electrohydrodynamic Atomization (EHDA)Document15 pagesElectrohydrodynamic Atomization (EHDA)Ananya SinghNo ratings yet
- Higher Education Catalogue 2017Document59 pagesHigher Education Catalogue 2017AtifNazNo ratings yet
- Bevel Gear DRAWING PDFDocument1 pageBevel Gear DRAWING PDFADITYA MOTLANo ratings yet
- Thesis Defense AljaberiDocument46 pagesThesis Defense AljaberiJose TamayoNo ratings yet
- Opera Rus MandrivaDocument118 pagesOpera Rus Mandrivaapi-3837985No ratings yet
- cs4l 2 0 en PDFDocument84 pagescs4l 2 0 en PDFmishka123No ratings yet
- Igcse Topic 1 Lesson 1 Water Cycle IgcseDocument25 pagesIgcse Topic 1 Lesson 1 Water Cycle Igcsedanielphilip68No ratings yet
- For The Sidereal Zodiac - Kenneth BowserDocument3 pagesFor The Sidereal Zodiac - Kenneth BowserGuilherme Alves PereiraNo ratings yet
- Hosford W.F. Materials For Engineers (CUP, 2008) (ISBN 9780521899970) (O) (298s) - EMDocument298 pagesHosford W.F. Materials For Engineers (CUP, 2008) (ISBN 9780521899970) (O) (298s) - EMZeeshan AliNo ratings yet
- Reading AdvocacyDocument1 pageReading AdvocacySamantha Jean PerezNo ratings yet
- Ushvinder Kaur - SSN PDFDocument48 pagesUshvinder Kaur - SSN PDFShraddha ShaduNo ratings yet
- TQM MCQ'S FOR SECOND MID-TERMDocument4 pagesTQM MCQ'S FOR SECOND MID-TERM17-550 Priyanka MaraboinaNo ratings yet
- Raiseplus Weekly Plan English Subject - Sample Only Region 5Document4 pagesRaiseplus Weekly Plan English Subject - Sample Only Region 5Jenelyn Ramos SantiagoNo ratings yet
- Forces and Motion 1Document5 pagesForces and Motion 1api-255910104No ratings yet
- Additional C QuestionsDocument41 pagesAdditional C Questionsvarikuti kalyaniNo ratings yet
- The Right To A Balanced and Healthful Ecology by Antonio G.M. La ViñaDocument30 pagesThe Right To A Balanced and Healthful Ecology by Antonio G.M. La Viñaellen joy chanNo ratings yet
- Introduction To Political PhilosophyDocument6 pagesIntroduction To Political PhilosophyMartina MartyNo ratings yet
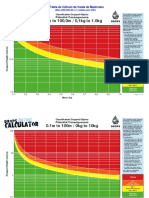
- MAM INS 30.4 Tabla Calculo Caida de MaterialesDocument2 pagesMAM INS 30.4 Tabla Calculo Caida de Materialeslourdes Tapia AlfaroNo ratings yet
- Working platforms for tracked plant designDocument16 pagesWorking platforms for tracked plant designLeandroNo ratings yet
- Gen Ed Day 5Document5 pagesGen Ed Day 5Jessica Villanueva100% (1)
- Call For Contributions - 2018 ATLAS T3 International Conference Babeș-Bolyai University, Cluj-Napoca, Romania, June 3-7, 2018Document16 pagesCall For Contributions - 2018 ATLAS T3 International Conference Babeș-Bolyai University, Cluj-Napoca, Romania, June 3-7, 2018Basarab Nicolescu100% (3)