You might also like
- Hemostasis Without Clot FormationDocument34 pagesHemostasis Without Clot FormationMiriam AguilarNo ratings yet
- Chronic Myeloid LeukemiaDocument8 pagesChronic Myeloid LeukemiaMiriam AguilarNo ratings yet
- Wms Main Pocket Guide 2017Document29 pagesWms Main Pocket Guide 2017justrudinNo ratings yet
- Indications For and Adverse Effects of Red-Cell TransfusionDocument12 pagesIndications For and Adverse Effects of Red-Cell TransfusionRoberto López Mata100% (1)
- Acute Lymphoblastic LeukemiaDocument9 pagesAcute Lymphoblastic LeukemiaMiriam AguilarNo ratings yet
- Myeloid LeukemiaDocument8 pagesMyeloid LeukemiaMiriam AguilarNo ratings yet
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Novel p53 Splicing Site Mutation in Li-Fraumeni-like Syndrome With OsteosarcomaDocument5 pagesNovel p53 Splicing Site Mutation in Li-Fraumeni-like Syndrome With OsteosarcomaMissy MishaNo ratings yet
- About Myelodysplastic Syndromes: Overview and TypesDocument12 pagesAbout Myelodysplastic Syndromes: Overview and TypesAulya ArchuletaNo ratings yet
- FGFGFGDocument1,196 pagesFGFGFGUyetBuyuNo ratings yet
- Histiocytic SarcomaDocument8 pagesHistiocytic SarcomadanishNo ratings yet
- Doxotil BrochurDocument2 pagesDoxotil BrochurAnonymous cxfknMNo ratings yet
- Community Health Survey: Assessment of Resident Health Needs and ConcernsDocument40 pagesCommunity Health Survey: Assessment of Resident Health Needs and ConcernsPreeti SachanNo ratings yet
- Cancer Treatment TypesDocument43 pagesCancer Treatment TypesM Sai Pradhyumna100% (2)
- Regimen Calgb 8811Document14 pagesRegimen Calgb 8811ricardo_cheNo ratings yet
- Genome Poster 2009Document1 pageGenome Poster 2009Öznur ErdoğanNo ratings yet
- Joseph LAWSON Tammy Malatak, On Behalf of Minor Child Elena LAWSON v. FORTIS INSURANCE COMPANY, Appellant/Cross-AppelleeDocument10 pagesJoseph LAWSON Tammy Malatak, On Behalf of Minor Child Elena LAWSON v. FORTIS INSURANCE COMPANY, Appellant/Cross-AppelleeScribd Government DocsNo ratings yet
- Malignant Diseases and Immunosuppression DrugsDocument36 pagesMalignant Diseases and Immunosuppression DrugsleonNo ratings yet
- Approach To The Adult With Pancytopenia - UpToDateDocument32 pagesApproach To The Adult With Pancytopenia - UpToDatePatricio RamirezNo ratings yet
- Lymphomas and LeukemiasDocument27 pagesLymphomas and LeukemiasgraceNo ratings yet
- Icnirp GuidelinesDocument20 pagesIcnirp Guidelinespuljke100% (1)
- Too Much of A Good Thing by Lee GoldmanDocument8 pagesToo Much of A Good Thing by Lee GoldmansimasNo ratings yet
- Child Health NSG Important QuestionsDocument10 pagesChild Health NSG Important Questionsedrinsne100% (2)
- Functions of Blood and ComponentsDocument15 pagesFunctions of Blood and ComponentsayuNo ratings yet
- Seminar On Cancer: Submitted To Submitted byDocument13 pagesSeminar On Cancer: Submitted To Submitted byUdaya SreeNo ratings yet
- Hematopoietic and Lymphoid Disorders GuideDocument49 pagesHematopoietic and Lymphoid Disorders GuideAbbi Yanto ArtNo ratings yet
- DiphtheriaDocument6 pagesDiphtheriaSigit Dwi RahardjoNo ratings yet
- 2010 Pediatrics 44Document21 pages2010 Pediatrics 44QueenOfCookies100% (2)
- MRCP 2 Practice Questions Book.1Document161 pagesMRCP 2 Practice Questions Book.1iban100% (2)
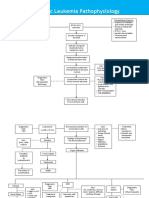
- Acute Lymphoblastic Leukemia Pathophysiology: Predisposing Factors: Etiology: Precipitating FactorsDocument3 pagesAcute Lymphoblastic Leukemia Pathophysiology: Predisposing Factors: Etiology: Precipitating FactorsKyla ValenciaNo ratings yet
- Vet Comparative Oncology - 2022 - Harris - Using Digital RNA Counting To Establish Flow Cytometry Diagnostic Criteria ForDocument10 pagesVet Comparative Oncology - 2022 - Harris - Using Digital RNA Counting To Establish Flow Cytometry Diagnostic Criteria Formarta idziakNo ratings yet
- Bone Marrow Biopsy Interpretive GuidelinesDocument19 pagesBone Marrow Biopsy Interpretive Guidelinesdtoxic1No ratings yet
- Disorders of White Blood Cells and Lymphoid TissuesDocument30 pagesDisorders of White Blood Cells and Lymphoid Tissuesammar amerNo ratings yet
- Soebandiri Division of Hematology & Medic Oncology Departement of Medicine Airlangga University of MedicineDocument23 pagesSoebandiri Division of Hematology & Medic Oncology Departement of Medicine Airlangga University of MedicineLaura ChandraNo ratings yet
- Rare Diseases 2013 PDFDocument56 pagesRare Diseases 2013 PDFantikenomaNo ratings yet
- Leukemia Cutis A Sign of Relapse.12Document3 pagesLeukemia Cutis A Sign of Relapse.12fatima amaliaNo ratings yet
- Blasts - Myeloid A1 en 01Document1 pageBlasts - Myeloid A1 en 01Gregorio De Las CasasNo ratings yet