You might also like
- Capítulo 13 Robbins PatologiaDocument7 pagesCapítulo 13 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 15 Robbins PatologiaDocument7 pagesCapítulo 15 Robbins PatologiaCarolina Stronger0% (1)
- Patologia - Resumo Robbins 28 - Sistema Nervoso Central NormalDocument3 pagesPatologia - Resumo Robbins 28 - Sistema Nervoso Central NormalPeperoniCourgueteNo ratings yet
- Capítulo 20 Robbins PatologiaDocument8 pagesCapítulo 20 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 17 Robbins PatologiaDocument8 pagesCapítulo 17 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 18 Robbins PatologiaDocument7 pagesCapítulo 18 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 10 Robbins PatologiaDocument8 pagesCapítulo 10 Robbins PatologiaCarolina StrongerNo ratings yet
- Tricuríase e EnterobioseDocument5 pagesTricuríase e EnterobioseMilhoeNo ratings yet
- Capítulo 11 Robbins PatologiaDocument8 pagesCapítulo 11 Robbins PatologiaCarolina StrongerNo ratings yet
- Ascaris, Trichuris, Enterobiose 2018Document37 pagesAscaris, Trichuris, Enterobiose 2018Sandy LorrayneNo ratings yet
- Capítulo 5 Robbins PatologiaDocument9 pagesCapítulo 5 Robbins PatologiaCarolina StrongerNo ratings yet
- Imunologia ClínicaDocument6 pagesImunologia ClínicaAna Carolina LopesNo ratings yet
- HematopoieseDocument61 pagesHematopoiesevannessa22No ratings yet
- #Respostas Dos Casos Clínicos de Microscopia.Document126 pages#Respostas Dos Casos Clínicos de Microscopia.OFF14No ratings yet
- Hematologia T3Document40 pagesHematologia T3Anonymous jmbY8AaJNo ratings yet
- Patologia - Resumo Robbins 1 - Adaptação, Dano e Morte CelularDocument11 pagesPatologia - Resumo Robbins 1 - Adaptação, Dano e Morte Celularwiller cassiano100% (2)
- Morfologia Do PlasmodiumDocument2 pagesMorfologia Do PlasmodiumJinc89100% (1)
- ImunologiaDocument10 pagesImunologiapiusinhaPIUSAO100% (2)
- Atlas de HematologiaDocument12 pagesAtlas de Hematologiagabrielle castroNo ratings yet
- Slides de Aula Unidade IDocument70 pagesSlides de Aula Unidade IFrantieskoNo ratings yet
- Capítulo 4 Robbins PatologiaDocument14 pagesCapítulo 4 Robbins PatologiaCarolina StrongerNo ratings yet
- Lúpus Eritematoso SistêmicoDocument21 pagesLúpus Eritematoso SistêmicorafaelNo ratings yet
- Estudo Dirigido Disturbio HemodinamicosDocument9 pagesEstudo Dirigido Disturbio HemodinamicosJuliana LimaNo ratings yet
- Granulopoese 2019.2 PDFDocument55 pagesGranulopoese 2019.2 PDFrobertovv100% (1)
- Apg 21 Soi IiiDocument8 pagesApg 21 Soi IiiYasmim Bastos Murta FloresNo ratings yet
- Caso ClínicoDocument2 pagesCaso ClínicoAmanda Medeiros100% (1)
- Capítulo 8 Robbins PatologiaDocument19 pagesCapítulo 8 Robbins PatologiaCarolina StrongerNo ratings yet
- Semiologia HematológicaDocument12 pagesSemiologia HematológicaLaiana Miralha CocaNo ratings yet
- Hematologia Basica Parte 2 - Composição Do SangueDocument21 pagesHematologia Basica Parte 2 - Composição Do SangueP2P3ESTATISTICA CIPTURNo ratings yet
- Estudo Dirigido - Inflamação e ReparoDocument9 pagesEstudo Dirigido - Inflamação e ReparoNatália MeloNo ratings yet
- Lupus Eritematoso SistemicoDocument15 pagesLupus Eritematoso Sistemicosharon barrosNo ratings yet
- Aula 10 HipersensibildadeDocument63 pagesAula 10 HipersensibildadeMaryB.HbrandtNo ratings yet
- Protozoários e ProtozoosesDocument3 pagesProtozoários e ProtozoosesMarianasouza Souza100% (1)
- Hemostasia e Coagulação SanguíneaDocument9 pagesHemostasia e Coagulação SanguíneaIngrid MarquesNo ratings yet
- Patologia - Resumo Robbins 4 - Disfunções Hemodinâmicas, Doenças Tromboembólica e ChoqueDocument6 pagesPatologia - Resumo Robbins 4 - Disfunções Hemodinâmicas, Doenças Tromboembólica e ChoqueKelly Cristina0% (2)
- Capítulo 3 Robbins PatologiaDocument8 pagesCapítulo 3 Robbins PatologiaCarolina StrongerNo ratings yet
- 3-Leishmania Velho MundoDocument70 pages3-Leishmania Velho MundoMayara StefanyNo ratings yet
- Anatomia Patológica SistemáticaDocument99 pagesAnatomia Patológica SistemáticaMargarida Sequeira MochoNo ratings yet
- Doenças Mielo e LinfoproliferativasDocument62 pagesDoenças Mielo e LinfoproliferativasbiabessaNo ratings yet
- Fisiologia de VinuezaDocument90 pagesFisiologia de VinuezaMarcelo PicoNo ratings yet
- MecanismoDocument17 pagesMecanismoLuana SouzaNo ratings yet
- Microbiologia e Imunologia - 20211.a - Aol 4Document3 pagesMicrobiologia e Imunologia - 20211.a - Aol 4LEONARDO MARTINSNo ratings yet
- Hemostasia e CoagulogramaDocument12 pagesHemostasia e Coagulogramasarahmili100% (1)
- Síndromes e Transtornos HematológicosDocument82 pagesSíndromes e Transtornos HematológicoseliasNo ratings yet
- 05 Artigo Exame PapanicolauDocument7 pages05 Artigo Exame PapanicolauJuliana RodriguesNo ratings yet
- Microbiologia Clinica Resumo 2Document7 pagesMicrobiologia Clinica Resumo 2soares9655100% (1)
- Aula 2 - BacteriasDocument37 pagesAula 2 - Bacteriasferreirasilvaa351No ratings yet
- Hematologia IiDocument34 pagesHematologia Iijosemar soaresNo ratings yet
- Capítulo 2 Robbins PatologiaDocument13 pagesCapítulo 2 Robbins PatologiaCarolina StrongerNo ratings yet
- BacillusDocument13 pagesBacillusAnia RobimNo ratings yet
- Perguntas de Patologia Que Eu LembroDocument3 pagesPerguntas de Patologia Que Eu LembroVictoria Pagung0% (1)
- Cap 13 Imunologia AbbasDocument2 pagesCap 13 Imunologia AbbasMatheusNo ratings yet
- Leucemia Mieloide CronicaDocument9 pagesLeucemia Mieloide CronicaAntonio CafezeiroNo ratings yet
- Apoptose - SlidesDocument56 pagesApoptose - SlidesJéssica Vaz0% (1)
- Núcleo, Diferenciação e Morte CelularDocument69 pagesNúcleo, Diferenciação e Morte CelularAmanda CristinaNo ratings yet
- Seminário - HemopatiasDocument77 pagesSeminário - HemopatiasMichely SantosNo ratings yet
- Guiao BGDocument5 pagesGuiao BGAna J SeabraNo ratings yet
- Genética Humana (Completa!!!)Document186 pagesGenética Humana (Completa!!!)Viviane Medeiros Silveira100% (2)
- GeneticaDocument58 pagesGeneticaIzabella PassamaniNo ratings yet
- Síndromes Hipertensivas Na GestaçãoDocument3 pagesSíndromes Hipertensivas Na GestaçãoCarolina StrongerNo ratings yet
- Síndromes Hemorrágicas Da 1 Metade Da GestaçãoDocument5 pagesSíndromes Hemorrágicas Da 1 Metade Da GestaçãoCarolina StrongerNo ratings yet
- Ebook AnestesiologiaDocument38 pagesEbook AnestesiologiaCarolina StrongerNo ratings yet
- Capítulo 17 Robbins PatologiaDocument8 pagesCapítulo 17 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 18 Robbins PatologiaDocument7 pagesCapítulo 18 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 9 Robbins PatologiaDocument12 pagesCapítulo 9 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 11 Robbins PatologiaDocument8 pagesCapítulo 11 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 14 Robbins PatologiaDocument3 pagesCapítulo 14 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 10 Robbins PatologiaDocument8 pagesCapítulo 10 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 8 Robbins PatologiaDocument19 pagesCapítulo 8 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 5 Robbins PatologiaDocument9 pagesCapítulo 5 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 4 Robbins PatologiaDocument14 pagesCapítulo 4 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 2 Robbins PatologiaDocument13 pagesCapítulo 2 Robbins PatologiaCarolina StrongerNo ratings yet
- Capítulo 3 Robbins PatologiaDocument8 pagesCapítulo 3 Robbins PatologiaCarolina StrongerNo ratings yet
- Historico 202111700447 PDFDocument5 pagesHistorico 202111700447 PDFana cecilia nascimentoNo ratings yet
- Anexo 2 Curso Educação AmbientalDocument4 pagesAnexo 2 Curso Educação AmbientalLurdinha NunesNo ratings yet
- Edital PAIC 2024 2025 FMT HVD CORRETODocument9 pagesEdital PAIC 2024 2025 FMT HVD CORRETOEvandro Portela CarneiroNo ratings yet
- Avaliação de Pesquisa - Base Histórica e Legal Da EducaçãoDocument5 pagesAvaliação de Pesquisa - Base Histórica e Legal Da Educaçãolillianfran6No ratings yet
- SOCIOLINGUISTICADocument8 pagesSOCIOLINGUISTICARigoberto Espinosa100% (3)
- Kit de ReceitasDocument27 pagesKit de ReceitasAna Paula de OliveiraNo ratings yet
- Curriculo Edson GonçalvesDocument2 pagesCurriculo Edson GonçalvesEdson GonçalvesNo ratings yet
- Apostila Transtornos de Aprendizagem X DesenvolvimDocument23 pagesApostila Transtornos de Aprendizagem X DesenvolvimPatriciaNo ratings yet
- Course NJ F8 MTYy MQDocument1 pageCourse NJ F8 MTYy MQFelipe LucasNo ratings yet
- A Sexualização de Crianças e Adolescentes Na MídiaDocument11 pagesA Sexualização de Crianças e Adolescentes Na MídiaMaria Tereza Buss WesslerNo ratings yet
- Edital-032022-Inscricoes-Homologadaspos-recurso - 3 2Document13 pagesEdital-032022-Inscricoes-Homologadaspos-recurso - 3 2tbhvp7sgchNo ratings yet
- Seis Regras para PersuadirDocument2 pagesSeis Regras para PersuadirGinamagalhãesNo ratings yet
- Cópia de Atividade Avaliativa Da Unidade IIDocument5 pagesCópia de Atividade Avaliativa Da Unidade IIWadna AmorNo ratings yet
- Metodologia Do Ensino Da Matematica UnoparDocument196 pagesMetodologia Do Ensino Da Matematica UnoparThiago Laurindo 2100% (1)
- Relatório Do Projeto de Leitura 2017 - Versão FinalDocument4 pagesRelatório Do Projeto de Leitura 2017 - Versão FinalAnderson KerllyNo ratings yet
- Políticas Curriculares: Entre o Bacalhau e A Feijoada!: Meta Da AulaDocument13 pagesPolíticas Curriculares: Entre o Bacalhau e A Feijoada!: Meta Da Aulavalnei de andradeNo ratings yet
- Programas Do 2º Ciclo Do ENSINO PRIMARIODocument263 pagesProgramas Do 2º Ciclo Do ENSINO PRIMARIOamiltonNo ratings yet
- Diversidade e ComunidadeDocument14 pagesDiversidade e ComunidadeANNA PAULA AIRES DE SOUZANo ratings yet
- v.3, n.5, 2009Document264 pagesv.3, n.5, 2009ifuanduNo ratings yet
- ProfileDocument4 pagesProfileAna Carla Castilho PagliocoNo ratings yet
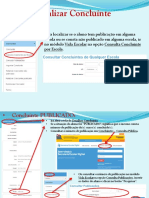
- Tutorial Localizao Migrao Editar Excluir Cancelar Seleo e Validao de Concluinte Na Sed PDFDocument20 pagesTutorial Localizao Migrao Editar Excluir Cancelar Seleo e Validao de Concluinte Na Sed PDFPaulo OliveiraNo ratings yet
- Ebook - INTERPRETACAO DE TEMA DE REDAÇÃO PROTAGONISTA OKDocument38 pagesEbook - INTERPRETACAO DE TEMA DE REDAÇÃO PROTAGONISTA OKWilza LimaNo ratings yet
- VULNERABILIDADES, GESTÃO DE SEGURANÇA PÚBLICA E CIDADES: o Papel Dos Municípios No Combate Às ViolênciaDocument16 pagesVULNERABILIDADES, GESTÃO DE SEGURANÇA PÚBLICA E CIDADES: o Papel Dos Municípios No Combate Às ViolênciaCLAUDIO ALBERTO GABRIEL GUIMARÃESNo ratings yet
- A Possessao e A Construcao Ritual Da PessoaDocument211 pagesA Possessao e A Construcao Ritual Da PessoaronaldotrindadeNo ratings yet
- MX5 Avaliacao Outubro2023 VersaoAlunoDocument4 pagesMX5 Avaliacao Outubro2023 VersaoAlunocarlosferreirajorgeNo ratings yet
- Concreto Auto AdensávelDocument5 pagesConcreto Auto AdensávelMatheusM.FerreiraNo ratings yet
- 03 Bases NeurocientificasDocument160 pages03 Bases NeurocientificasAna Paula SantosNo ratings yet
- Programacao 2023 V2 Programacao GeralDocument33 pagesProgramacao 2023 V2 Programacao GeralAngelo AguirreNo ratings yet
- AVA 2 - Organização Da Educação EscolarDocument5 pagesAVA 2 - Organização Da Educação EscolarBeatriz BragaNo ratings yet
- PROLICEN - VII Congresso de Pesquisa Ensino e Extensao-Conhecimento e Desenvolvimento SustentavelDocument536 pagesPROLICEN - VII Congresso de Pesquisa Ensino e Extensao-Conhecimento e Desenvolvimento SustentavelJuliana Moral PereiraNo ratings yet