You might also like
- GeneticsDocument157 pagesGeneticsShruti TiwariNo ratings yet
- Class - X - Science - Heredity and Evolution - PPT PDFDocument11 pagesClass - X - Science - Heredity and Evolution - PPT PDFAvrian PradiptyaNo ratings yet
- HeredityDocument4 pagesHereditysamyukthaNo ratings yet
- Genetics in Orthodontics - 12 11 23Document88 pagesGenetics in Orthodontics - 12 11 23dr.sakshigarg0105No ratings yet
- HEREDITYDocument14 pagesHEREDITYGt FansNo ratings yet
- Epi - (Genetic Epidemiology Mam Slides)Document167 pagesEpi - (Genetic Epidemiology Mam Slides)Naga dharshiniNo ratings yet
- 8 GeneticsDocument85 pages8 GeneticsRoselyn CarmenNo ratings yet
- BIO301 Handouts 1 167Document351 pagesBIO301 Handouts 1 167Tayyaba ArbabNo ratings yet
- Heredity and Evolution Class 10 Notes CBSE Science Chapter 9 PDFDocument11 pagesHeredity and Evolution Class 10 Notes CBSE Science Chapter 9 PDFvanshNo ratings yet
- CL 12 Bio Chapter-5 Principle of Inheritance and VariationDocument53 pagesCL 12 Bio Chapter-5 Principle of Inheritance and Variationjackieaj093No ratings yet
- Genetics in OrthodonticsDocument63 pagesGenetics in OrthodonticsPrachi Madan100% (20)
- Lesson 1-2 Genbio2Document5 pagesLesson 1-2 Genbio2yeoflittlefaithNo ratings yet
- Mendelian Genetics ExplainedDocument53 pagesMendelian Genetics Explainedlaica andalNo ratings yet
- CH 5 BIODocument9 pagesCH 5 BIOKrish YadavNo ratings yet
- Ch. 23 Lecture Outline PDFDocument15 pagesCh. 23 Lecture Outline PDFCSERM UNASNo ratings yet
- Definition of Gene (Repaired)Document107 pagesDefinition of Gene (Repaired)Ananda PreethiNo ratings yet
- Notes - Heredity and EvolutionDocument16 pagesNotes - Heredity and EvolutionweirdoNo ratings yet
- Genetics Study GuideDocument7 pagesGenetics Study GuideAanav MittalNo ratings yet
- Mendelian GeneticsDocument6 pagesMendelian GeneticsMaria Danica100% (2)
- Mitosis and Meiosis DivisionDocument3 pagesMitosis and Meiosis Divisionrubab azizNo ratings yet
- Genetics Complete NotesDocument71 pagesGenetics Complete NotesSmritiNo ratings yet
- Mendelian and Population GeneticsDocument30 pagesMendelian and Population GeneticsJuniaNo ratings yet
- ACFrOgAfRr3sSsuCAkrajXlVJlNW9CEtIjaaAShbzz53F - BV5yV6DzQdxtAJFRZXXlCzNJ2z436IplXhlNZcLstm s3JGzE2BiehEN5Wj1zYDQg1luLD89ztag DeLdXlbq5Nbv38FIU g05ICjDocument32 pagesACFrOgAfRr3sSsuCAkrajXlVJlNW9CEtIjaaAShbzz53F - BV5yV6DzQdxtAJFRZXXlCzNJ2z436IplXhlNZcLstm s3JGzE2BiehEN5Wj1zYDQg1luLD89ztag DeLdXlbq5Nbv38FIU g05ICjCarl SeanNo ratings yet
- Study Materials: Vedantu Innovations Pvt. Ltd. Score High With A Personal Teacher, Learn LIVE Online!Document8 pagesStudy Materials: Vedantu Innovations Pvt. Ltd. Score High With A Personal Teacher, Learn LIVE Online!sushantNo ratings yet
- Biology of HeredityDocument5 pagesBiology of Hereditypalmer okiemuteNo ratings yet
- Analyzing Earlobe Traits in a Family PedigreeDocument21 pagesAnalyzing Earlobe Traits in a Family PedigreeAnonymous DWvfJuMcQNo ratings yet
- MonohybridGeneticsPract1Document8 pagesMonohybridGeneticsPract1Muhammad AwaisNo ratings yet
- Screenshot 2022-11-29 at 6.08.24 PMDocument34 pagesScreenshot 2022-11-29 at 6.08.24 PMpandiyammalkrishnanNo ratings yet
- Genetics and Consequences of DomesticationDocument10 pagesGenetics and Consequences of DomesticationChandra Sekhar KaranaNo ratings yet
- LV (Lyu) Hexin (吕和鑫), Ph.D. Associate Professor Office: 2#-107 Wechat/Mobile:86-15822765529 Email: lvhx@tust.edu.cnDocument61 pagesLV (Lyu) Hexin (吕和鑫), Ph.D. Associate Professor Office: 2#-107 Wechat/Mobile:86-15822765529 Email: lvhx@tust.edu.cncharith chiranthaNo ratings yet
- Biol 12 Genetics Notes and University Entrance Exam Questions 2003Document66 pagesBiol 12 Genetics Notes and University Entrance Exam Questions 2003Tizita EmanaNo ratings yet
- Principles of Genetics: Module - 3Document26 pagesPrinciples of Genetics: Module - 3Indrojyoti MondalNo ratings yet
- Genetics and Orthodontics-FinalDocument96 pagesGenetics and Orthodontics-FinalRaj SinghNo ratings yet
- Genetics and Heredity Margie de LeonDocument56 pagesGenetics and Heredity Margie de LeonCarmsNo ratings yet
- Heredity and Evolution NotesDocument5 pagesHeredity and Evolution NotesCapt Pradeep kumarNo ratings yet
- Plant Genetics: Group 10 MembersDocument25 pagesPlant Genetics: Group 10 MembersJhemaeca PalisocNo ratings yet
- GeneticsDocument10 pagesGeneticsRivisha SiriwardanaNo ratings yet
- JojoDocument3 pagesJojoHui Robert ChengNo ratings yet
- Variation F 5Document8 pagesVariation F 5Angie Kong Su MeiNo ratings yet
- Inheritance and Variation Notes for Class XII BiologyDocument9 pagesInheritance and Variation Notes for Class XII BiologyAshok KumarNo ratings yet
- CBSE Class 10 Science Notes Chapter 9 Heredity and EvolutionDocument18 pagesCBSE Class 10 Science Notes Chapter 9 Heredity and EvolutionHarsh AgarwalNo ratings yet
- Mendelian GeneticsDocument49 pagesMendelian GeneticsKaiye ReitarNo ratings yet
- Genetics: Inheritance Refers To The Process by Which Characters/traits Are Passed FromDocument55 pagesGenetics: Inheritance Refers To The Process by Which Characters/traits Are Passed FromTop gadgets and moviesNo ratings yet
- Biology Notes F4Document59 pagesBiology Notes F4Nathan SsekamatteNo ratings yet
- Principles of GeneticsDocument26 pagesPrinciples of GeneticsKofoworola MikailNo ratings yet
- CH 23 Lecture OutlineDocument6 pagesCH 23 Lecture OutlineZohal Ghulam-JelaniNo ratings yet
- Bio502 Handouts 1 50.pin2Document156 pagesBio502 Handouts 1 50.pin2Mohammadihsan NoorNo ratings yet
- Genetics BookDocument77 pagesGenetics BookelctoyasNo ratings yet
- Heredity and VariationDocument25 pagesHeredity and VariationDre ParkerNo ratings yet
- Basic Principles of GeneticsDocument3 pagesBasic Principles of GeneticsKuoRosalineNo ratings yet
- Biology Project (Pedigree Analysis)Document24 pagesBiology Project (Pedigree Analysis)vivekmangla06No ratings yet
- BIO 101 Notes-3Document12 pagesBIO 101 Notes-3fmukuka12No ratings yet
- Heredity Class 10Document40 pagesHeredity Class 10ChaithraNo ratings yet
- GeneticsDocument6 pagesGeneticsSamyak JainNo ratings yet
- GENETICSDocument6 pagesGENETICSSamyak JainNo ratings yet
- Genetics in OrthodonticsDocument107 pagesGenetics in OrthodonticsdrgreeshmahariniNo ratings yet
- Class-10 Notes CH-9 Heredity and EvolutionDocument13 pagesClass-10 Notes CH-9 Heredity and EvolutionN.R. ShirishaNo ratings yet
- Summary of The Gene: by Siddhartha Mukherjee | Includes AnalysisFrom EverandSummary of The Gene: by Siddhartha Mukherjee | Includes AnalysisNo ratings yet
- Gen PTL (MM) NPT % Pollen Stainability of Pollen (IKI) (Mean Sem) (Mean Sem) Germ %viable %non-ViableDocument5 pagesGen PTL (MM) NPT % Pollen Stainability of Pollen (IKI) (Mean Sem) (Mean Sem) Germ %viable %non-ViableTeflon SlimNo ratings yet
- R Codes Germinate Seed Data AnalysisDocument1 pageR Codes Germinate Seed Data AnalysisTeflon SlimNo ratings yet
- Results FileDocument159 pagesResults FileTeflon SlimNo ratings yet
- Combined Diallel-W DataDocument703 pagesCombined Diallel-W DataTeflon SlimNo ratings yet
- File Name: M0928 Location: Abuja PLO: Rep Entry Pedigree Yieldin Yieldu Plstin Plstun Dyskin DyskunDocument48 pagesFile Name: M0928 Location: Abuja PLO: Rep Entry Pedigree Yieldin Yieldu Plstin Plstun Dyskin DyskunTeflon SlimNo ratings yet
- Additive NonadditiveDocument1 pageAdditive NonadditiveTeflon SlimNo ratings yet
- Ideal Tester STR-BA1057Document7 pagesIdeal Tester STR-BA1057Teflon SlimNo ratings yet
- Interploidy Hybridization in Sympatric Zones The FormationDocument14 pagesInterploidy Hybridization in Sympatric Zones The FormationTeflon SlimNo ratings yet
- Regional Hybrid TrialDocument8 pagesRegional Hybrid TrialTeflon SlimNo ratings yet
- Hybrid Maize Yield and Trait DataDocument46 pagesHybrid Maize Yield and Trait DataTeflon SlimNo ratings yet
- Combining Ability of Extra-Early White Inbreds Final DraftDocument57 pagesCombining Ability of Extra-Early White Inbreds Final DraftTeflon SlimNo ratings yet
- GGE Biplot of GCA Stability AnalysisDocument24 pagesGGE Biplot of GCA Stability AnalysisTeflon SlimNo ratings yet
- SCA Effects of Grain Yield in Maize HybridsDocument1 pageSCA Effects of Grain Yield in Maize HybridsTeflon SlimNo ratings yet
- Obs Source Ss DF Ms Fvalue Probf 1 2 3 4: YieldDocument292 pagesObs Source Ss DF Ms Fvalue Probf 1 2 3 4: YieldTeflon SlimNo ratings yet
- Inbred Lines Evaluated in Nigeria, 2011.: MaizeDocument1 pageInbred Lines Evaluated in Nigeria, 2011.: MaizeTeflon SlimNo ratings yet
- Table 2Document1 pageTable 2Teflon SlimNo ratings yet
- Table 1. Description of The 20 Yellow-Endosperm Entry Pedigree Reaction To Low-N Reaction To Striga-InfestationDocument1 pageTable 1. Description of The 20 Yellow-Endosperm Entry Pedigree Reaction To Low-N Reaction To Striga-InfestationTeflon SlimNo ratings yet
- Table 3. GCA Effects of Extra-Early Yellow Inbred Parents For Grain Yield and Other Agronomic Traits Evaluated Across Test Environments in 2011Document1 pageTable 3. GCA Effects of Extra-Early Yellow Inbred Parents For Grain Yield and Other Agronomic Traits Evaluated Across Test Environments in 2011Teflon SlimNo ratings yet
- Responses To Reviewers' Comments CommentDocument4 pagesResponses To Reviewers' Comments CommentTeflon SlimNo ratings yet
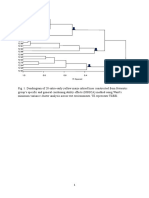
- Dendrogram of 20 extra-early yellow maize inbred lines constructed from HSGCA methodDocument1 pageDendrogram of 20 extra-early yellow maize inbred lines constructed from HSGCA methodTeflon SlimNo ratings yet
- Figure 1Document1 pageFigure 1Teflon SlimNo ratings yet
- Figure 5Document1 pageFigure 5Teflon SlimNo ratings yet
- Practical Session - Sample DataDocument21 pagesPractical Session - Sample DataTeflon SlimNo ratings yet
- Figure 4Document1 pageFigure 4Teflon SlimNo ratings yet
- Ba0727ik DataDocument12 pagesBa0727ik DataTeflon SlimNo ratings yet
- Anhu 12083Document8 pagesAnhu 12083Teflon SlimNo ratings yet
- Figure 2Document1 pageFigure 2Teflon SlimNo ratings yet
- Correlation and Regression Analyses: Measurement of More Than One Variable On Each Experimental or Observational UnitDocument23 pagesCorrelation and Regression Analyses: Measurement of More Than One Variable On Each Experimental or Observational UnitTeflon SlimNo ratings yet
- Data Analysis - Practical SessionDocument12 pagesData Analysis - Practical SessionTeflon SlimNo ratings yet
- Mathews Paul G. Design of Experiments With MINITAB PDFDocument521 pagesMathews Paul G. Design of Experiments With MINITAB PDFFernando Chavarria100% (2)
- Study of sugar cane bagasse hydrolysis using phosphoric acidDocument11 pagesStudy of sugar cane bagasse hydrolysis using phosphoric acidRené MartínezNo ratings yet
- Pharmaceutical Manufacturers Canada ListDocument3 pagesPharmaceutical Manufacturers Canada ListTaylor McBrideNo ratings yet
- Bayer CropScience submission to agricultural competitiveness white paperDocument27 pagesBayer CropScience submission to agricultural competitiveness white paperHamza ButtNo ratings yet
- Yeast Extract 212750Document2 pagesYeast Extract 212750Isna Echa AkiraNo ratings yet
- Recombinant DNA TechnologyDocument24 pagesRecombinant DNA TechnologySajjad AhmadNo ratings yet
- DisposableDocument9 pagesDisposableSuzanne TaylorNo ratings yet
- Assingment of BiochemicalDocument5 pagesAssingment of BiochemicalTalal AshrafNo ratings yet
- Ciba SandoDocument14 pagesCiba SandolmcNo ratings yet
- Enological Products and Their UsesDocument2 pagesEnological Products and Their UsespanospappasNo ratings yet
- Activation TaggingDocument10 pagesActivation TaggingprajaktabendreNo ratings yet
- Bacterial Expression Vectors - EMBLDocument9 pagesBacterial Expression Vectors - EMBLjvreddiNo ratings yet
- Poster PDFDocument1 pagePoster PDFAnonymous CPckXXNo ratings yet
- The International Journal of Biochemistry & Cell Biology Volume 76 Issue 2016 (Doi 10.1016/j.biocel.2016.04.014) Lofrumento, Dario Domenico La Piana, Gianluigi Palmitessa, Val - Stimulation by PRDocument7 pagesThe International Journal of Biochemistry & Cell Biology Volume 76 Issue 2016 (Doi 10.1016/j.biocel.2016.04.014) Lofrumento, Dario Domenico La Piana, Gianluigi Palmitessa, Val - Stimulation by PRWanabud abudNo ratings yet
- Noted Filipino scientists who contributed to biology, medicine, and agricultureDocument4 pagesNoted Filipino scientists who contributed to biology, medicine, and agriculturejuancho2020No ratings yet
- 27-Bacteria and ArchaeaDocument20 pages27-Bacteria and ArchaeaJaylan BrownNo ratings yet
- Biotech Production of Gluconic AcidDocument10 pagesBiotech Production of Gluconic AcidI. Murali KrishnaNo ratings yet
- Si RNADocument29 pagesSi RNALuis MonteroNo ratings yet
- Responsive Documents - CREW: FDA: Regarding FOIA Logs: 6/19/2012 - CREWLog ResponseDocument1,132 pagesResponsive Documents - CREW: FDA: Regarding FOIA Logs: 6/19/2012 - CREWLog ResponseCREWNo ratings yet
- Genetics Ninestiles TMIDDocument14 pagesGenetics Ninestiles TMIDPatrick AbidraNo ratings yet
- Chromosomal KaryotypesDocument12 pagesChromosomal KaryotypesCess DichosaNo ratings yet
- Epigenetics: - Epi'genetics - On' or Over' The Genetic Information Encoded in The DNADocument14 pagesEpigenetics: - Epi'genetics - On' or Over' The Genetic Information Encoded in The DNAlordniklausNo ratings yet
- 2014 Article 1340Document11 pages2014 Article 1340Karen Avalos VelaNo ratings yet
- Science Magazine 5724 2005 05 13Document167 pagesScience Magazine 5724 2005 05 13dragon*No ratings yet
- 8DT24 - and - 8DT24 - 2 DTDocument1 page8DT24 - and - 8DT24 - 2 DTRusu CiprianNo ratings yet
- Sales Bill Jan20Document74 pagesSales Bill Jan20Mansoor TheenNo ratings yet
- Biochem Nucleic Acid ReviewerDocument5 pagesBiochem Nucleic Acid ReviewerGabrielle FranciscoNo ratings yet
- Toxicity Testing in IndiaDocument31 pagesToxicity Testing in Indiavidya sagarNo ratings yet
- UPSC Zoology Syllabus - IAS Mains Optional SubjectsDocument5 pagesUPSC Zoology Syllabus - IAS Mains Optional SubjectsNAVEENNo ratings yet
- 3.4 Microbiology PDFDocument31 pages3.4 Microbiology PDFQueena Yan Yie LeeNo ratings yet
- Viktoria ItalijaDocument6 pagesViktoria ItalijaMilijana KovačevićNo ratings yet