You might also like
- Compendium of Atomic Alkali Resistant Optical Thin Films, Diffusion and Electrical Mobility in Diode Pumped Alkali Lasers (DPALs)From EverandCompendium of Atomic Alkali Resistant Optical Thin Films, Diffusion and Electrical Mobility in Diode Pumped Alkali Lasers (DPALs)No ratings yet
- Wo3 NanorodsDocument6 pagesWo3 NanorodsinfinitopNo ratings yet
- MPM144 13 SakharovDocument6 pagesMPM144 13 SakharovВадим АрыковNo ratings yet
- Msa20111000007 87029177Document5 pagesMsa20111000007 87029177Mario Misael Machado LòpezNo ratings yet
- Electrochromism of Sputtered Fluorinated Titanium Oxide Thin FilmsDocument4 pagesElectrochromism of Sputtered Fluorinated Titanium Oxide Thin FilmsTanveer ZiaNo ratings yet
- Electric Contacts For ZnODocument5 pagesElectric Contacts For ZnOjmcorreahNo ratings yet
- Lead Dioxide 4Document4 pagesLead Dioxide 4Khobaib HayatNo ratings yet
- Ag TiO2 SintesiDocument8 pagesAg TiO2 SintesiLuca AccorsiNo ratings yet
- Electrochemical Etching PDFDocument26 pagesElectrochemical Etching PDFMohamed ShaganNo ratings yet
- Photoelectrochemical Studies of Nanocrystalline Tio Film ElectrodesDocument8 pagesPhotoelectrochemical Studies of Nanocrystalline Tio Film ElectrodesFayeen K ShariarNo ratings yet
- Journal Pre-Proof: Thin Solid FilmsDocument31 pagesJournal Pre-Proof: Thin Solid FilmsSahin CoskunNo ratings yet
- Dilute Electrodeposition of TiO2 and ZnO Thin Film MemristorsDocument6 pagesDilute Electrodeposition of TiO2 and ZnO Thin Film Memristorsalagar krishna kumarNo ratings yet
- InTech-Chasing High Efficiency DSSC by Nano Structural Surface Engineering at Low Processing Temperature For Titanium Dioxide ElectrodesDocument19 pagesInTech-Chasing High Efficiency DSSC by Nano Structural Surface Engineering at Low Processing Temperature For Titanium Dioxide ElectrodesDeva RajNo ratings yet
- Accepted ManuscriptDocument22 pagesAccepted ManuscriptLoubna MentarNo ratings yet
- Plasmonic Gold Nanocrystals Coupled With Photonic CrystalDocument7 pagesPlasmonic Gold Nanocrystals Coupled With Photonic CrystalKatia ONo ratings yet
- Micro System Technologies 2008 VDocument9 pagesMicro System Technologies 2008 VOmar ReynosoNo ratings yet
- Enhancing The Performance of Dye-Sensitized Solar Cells Based On An Organic Dye by Incorporating TiO2 Nanotube in A TiO2 Nanoparticle FilmDocument8 pagesEnhancing The Performance of Dye-Sensitized Solar Cells Based On An Organic Dye by Incorporating TiO2 Nanotube in A TiO2 Nanoparticle FilmMuhammad Bilal QadirNo ratings yet
- Rose-Like Superhydrophobic Surface Based On Conducting Dmit SaltDocument5 pagesRose-Like Superhydrophobic Surface Based On Conducting Dmit SaltNguyễn Toàn ThắngNo ratings yet
- Buffer1 s2.0 S0272884213003519 Main PDFDocument7 pagesBuffer1 s2.0 S0272884213003519 Main PDFeid elsayedNo ratings yet
- Magnetic Conductive Outer Layer in Oxygen-Deficient Tio Single CrystalsDocument7 pagesMagnetic Conductive Outer Layer in Oxygen-Deficient Tio Single CrystalsSahil AkramNo ratings yet
- GoldCatalyst CrystallizationDocument6 pagesGoldCatalyst CrystallizationthuronNo ratings yet
- Synthesis and Deposition of Ag Nanoparticles by CoDocument9 pagesSynthesis and Deposition of Ag Nanoparticles by Comonikasharma1604No ratings yet
- 1996 J Chem Soc Ag RougheningDocument7 pages1996 J Chem Soc Ag RougheningSara AldabeNo ratings yet
- Electrodeposition of MetalDocument4 pagesElectrodeposition of MetalvkmsNo ratings yet
- 2 s2.0 85030846253Document7 pages2 s2.0 85030846253ARSALAN AHMADNo ratings yet
- Choosing the Right Transparent ConductorDocument6 pagesChoosing the Right Transparent ConductorronbinyeNo ratings yet
- Ijaiem 2014 04 26 057Document5 pagesIjaiem 2014 04 26 057International Journal of Application or Innovation in Engineering & ManagementNo ratings yet
- High Optical Transmittance of Nanostructured TiO2 Thin Film Using Dip Coating MethodDocument5 pagesHigh Optical Transmittance of Nanostructured TiO2 Thin Film Using Dip Coating MethodrangaanshumNo ratings yet
- Photocurrent Enhancement by Surface Plasmon Resonance of Silver Nanoparticles in Highly Porous Dye-Sensitized Solar CellsDocument6 pagesPhotocurrent Enhancement by Surface Plasmon Resonance of Silver Nanoparticles in Highly Porous Dye-Sensitized Solar CellsMuhammad Bilal QadirNo ratings yet
- SdarticleDocument6 pagesSdarticleBoodeppa NatarajuNo ratings yet
- 1-D ZnO Nanostructures for ETA Solar CellsDocument4 pages1-D ZnO Nanostructures for ETA Solar CellsTôn Vương NguyễnNo ratings yet
- Ijetr022687 PDFDocument4 pagesIjetr022687 PDFerpublicationNo ratings yet
- Synthesis of Zinc Oxide Nanocrystalline Powders For Cosmetic ApplicationsDocument6 pagesSynthesis of Zinc Oxide Nanocrystalline Powders For Cosmetic ApplicationsMaritnela Gomez JimenezNo ratings yet
- Thin Solid Films: Ching-Hsun Chao, Chi-Lung Chang, Chien-Hung Chan, Shui-Yang Lien, Ko-Wei Weng, Kuo-Shan YaoDocument4 pagesThin Solid Films: Ching-Hsun Chao, Chi-Lung Chang, Chien-Hung Chan, Shui-Yang Lien, Ko-Wei Weng, Kuo-Shan YaoTrio Yuda SeptiawanNo ratings yet
- Self-Aligned, Insulating-Layer Structure For Integrated Fabrication of Organic Self-Assembled Multilayer Electronic DevicesDocument4 pagesSelf-Aligned, Insulating-Layer Structure For Integrated Fabrication of Organic Self-Assembled Multilayer Electronic DevicesemediageNo ratings yet
- 847 AugustineCDocument10 pages847 AugustineCRenato EvangelistaNo ratings yet
- Published On Materials Chemistry and Physics Volume 129, Issues 1-2, 15 September 2011, Pages 553-557 Author ManuscriptDocument5 pagesPublished On Materials Chemistry and Physics Volume 129, Issues 1-2, 15 September 2011, Pages 553-557 Author Manuscriptvungau1992No ratings yet
- Selective Deposition of Insulating Metal Oxide in Perovskite Solar Cells With Enhanced Device PerformanceDocument14 pagesSelective Deposition of Insulating Metal Oxide in Perovskite Solar Cells With Enhanced Device Performanceadharsh27No ratings yet
- Material & Energy Balancing in Process IndustryDocument10 pagesMaterial & Energy Balancing in Process IndustryGeorge Michael Alvarado LopezNo ratings yet
- Smith Et Al-2011-Advanced MaterialsDocument5 pagesSmith Et Al-2011-Advanced MaterialsEkashmi RathoreNo ratings yet
- Growth of Passive Films On Valve Metals and Their AlloysDocument9 pagesGrowth of Passive Films On Valve Metals and Their AlloysDjedili AmelNo ratings yet
- Dye-Sensitized Solar Cell Using Local Chlorin Dye Achieves 1.00% EfficiencyDocument7 pagesDye-Sensitized Solar Cell Using Local Chlorin Dye Achieves 1.00% EfficiencyFisikaku IdolakuNo ratings yet
- Joy, Sep 2011Document4 pagesJoy, Sep 2011emediageNo ratings yet
- Articles: Photoluminescence Properties of Sno Nanoparticles Synthesized by Sol-Gel MethodDocument5 pagesArticles: Photoluminescence Properties of Sno Nanoparticles Synthesized by Sol-Gel MethodVikas PatilNo ratings yet
- Anodized Titanium Dioxide Films: Morphology, Wettability & PhotocurrentDocument7 pagesAnodized Titanium Dioxide Films: Morphology, Wettability & PhotocurrentRik ChatterjeeNo ratings yet
- Var Key 1993Document4 pagesVar Key 1993SoufianeBenhamidaNo ratings yet
- Effect of Electrodeposition Parameters On Morphology of Copper Thin FilmsDocument7 pagesEffect of Electrodeposition Parameters On Morphology of Copper Thin FilmsIOSRJEN : hard copy, certificates, Call for Papers 2013, publishing of journalNo ratings yet
- Lithium Acceptors and Hydrogen in Zno: Caleb Corolewski Rough DraftDocument20 pagesLithium Acceptors and Hydrogen in Zno: Caleb Corolewski Rough Draftlancealot24No ratings yet
- Controlled Electrodeposition of Nanoparticulate Tungsten OxideDocument4 pagesControlled Electrodeposition of Nanoparticulate Tungsten OxideemediageNo ratings yet
- Synthesis of Yellow Colloidal SilverDocument4 pagesSynthesis of Yellow Colloidal SilverOng Chin LengNo ratings yet
- EXPERIMENT 8. Monolayer Characterization: Contact Angles, Reflection Infrared Spectroscopy, and EllipsometryDocument9 pagesEXPERIMENT 8. Monolayer Characterization: Contact Angles, Reflection Infrared Spectroscopy, and EllipsometryavniNo ratings yet
- Photocatalytic Degradation of Methylene Blue On Nanocrystalline TiO2Document7 pagesPhotocatalytic Degradation of Methylene Blue On Nanocrystalline TiO2Michel G. RahalNo ratings yet
- Monolayer-Protected Metal Nanoparticle: Toluene) Auc L Toluene) N (C H Auc L N (C HDocument27 pagesMonolayer-Protected Metal Nanoparticle: Toluene) Auc L Toluene) N (C H Auc L N (C HmichsantosNo ratings yet
- Plasmonic Photo Catalyst For H2 Evolution in Photo Catalytic Water SplittingDocument7 pagesPlasmonic Photo Catalyst For H2 Evolution in Photo Catalytic Water Splittingbsnyder3No ratings yet
- ZnF2 PbO TeO2 TiO2Document8 pagesZnF2 PbO TeO2 TiO2hanumatharao kNo ratings yet
- I-V Characteristic of Cdo/Ps HeterojunctionDocument6 pagesI-V Characteristic of Cdo/Ps HeterojunctionInternational Journal of Application or Innovation in Engineering & ManagementNo ratings yet
- Comparative Study of Nanostructured TiO2, ZnO and Bilayer TiO2/ZnO Dye-Sensitized Solar CellsDocument9 pagesComparative Study of Nanostructured TiO2, ZnO and Bilayer TiO2/ZnO Dye-Sensitized Solar CellsbismuthsunilNo ratings yet
- Investigation of Photodeposition of Gold On Titanium Dioxide NanoparticlesDocument7 pagesInvestigation of Photodeposition of Gold On Titanium Dioxide NanoparticlesACHIENG REBECCANo ratings yet
- ZnO Coating Enhances Dye-Sensitized Solar Cell EfficiencyDocument6 pagesZnO Coating Enhances Dye-Sensitized Solar Cell EfficiencyIntenNo ratings yet
- აუტომოტივი, თებერვალი 2009Document100 pagesაუტომოტივი, თებერვალი 2009emediageNo ratings yet
- დიალოგი, ივნისი 2010Document76 pagesდიალოგი, ივნისი 2010emediageNo ratings yet
- აუტომოტივი, თებერვალი 2009Document100 pagesაუტომოტივი, თებერვალი 2009emediageNo ratings yet
- აუტომოტივი, თებერვალი 2009Document100 pagesაუტომოტივი, თებერვალი 2009emediageNo ratings yet
- დიალოგი, იანვარი 2012Document68 pagesდიალოგი, იანვარი 2012emediageNo ratings yet
- g, იანვარი 2012Document7 pagesg, იანვარი 2012emediageNo ratings yet
- აუტომოტივი, თებერვალი 2009Document100 pagesაუტომოტივი, თებერვალი 2009emediageNo ratings yet
- აუტომოტივი, თებერვალი 2009Document100 pagesაუტომოტივი, თებერვალი 2009emediageNo ratings yet
- აუტომოტივი, თებერვალი 2009Document100 pagesაუტომოტივი, თებერვალი 2009emediageNo ratings yet
- გაზეთი ქართული, მარტი 2011Document2 pagesგაზეთი ქართული, მარტი 2011emediageNo ratings yet
- Simple JAVA (Russian)Document231 pagesSimple JAVA (Russian)Dana DingaNo ratings yet
- დიალოგი, ივნისი 2010Document76 pagesდიალოგი, ივნისი 2010emediageNo ratings yet
- აუტომოტივი, თებერვალი 2009Document100 pagesაუტომოტივი, თებერვალი 2009emediageNo ratings yet
- Simple JAVA (Russian)Document231 pagesSimple JAVA (Russian)Dana DingaNo ratings yet
- მირიანა, თებერვალი 2012Document100 pagesმირიანა, თებერვალი 2012emediageNo ratings yet
- The First Magazine Test, თებერვალი 2012Document43 pagesThe First Magazine Test, თებერვალი 2012emediageNo ratings yet
- Css3 Cheat SheetDocument1 pageCss3 Cheat SheetPhilip SoaresNo ratings yet
- Fenvik, მარტი 2012Document7 pagesFenvik, მარტი 2012emediageNo ratings yet
- მირიანა, თებერვალი 2012Document100 pagesმირიანა, თებერვალი 2012emediageNo ratings yet
- მირიანა, თებერვალი 2012Document100 pagesმირიანა, თებერვალი 2012emediageNo ratings yet
- Fenvik, მარტი 2012Document180 pagesFenvik, მარტი 2012emediageNo ratings yet
- Fenvik, მარტი 2012Document180 pagesFenvik, მარტი 2012emediageNo ratings yet
- Fenvik, მარტი 2012Document50 pagesFenvik, მარტი 2012emediageNo ratings yet
- მირიანა, თებერვალი 2012Document100 pagesმირიანა, თებერვალი 2012emediageNo ratings yet
- You and Your Family, Oct 2011Document5 pagesYou and Your Family, Oct 2011emediageNo ratings yet
- დდდ, მარტი 2012Document4 pagesდდდ, მარტი 2012emediageNo ratings yet
- Awake!, Jul 2011Document4 pagesAwake!, Jul 2011emediageNo ratings yet
- You and Your Family, Jan 2011Document4 pagesYou and Your Family, Jan 2011emediageNo ratings yet
- You and Your Family, Apr 2011Document5 pagesYou and Your Family, Apr 2011emediageNo ratings yet
- Synthesis of Metal-Loaded Poly (Aminohexyl) (Aminopropyl) Silsesquioxane Colloids and Their Self-Organization Into DendritesDocument4 pagesSynthesis of Metal-Loaded Poly (Aminohexyl) (Aminopropyl) Silsesquioxane Colloids and Their Self-Organization Into DendritesemediageNo ratings yet
- 300G IM SettingsSheets 20160122Document27 pages300G IM SettingsSheets 20160122zeljkoradaNo ratings yet
- Spotlight Planet Magazine UpdatedDocument32 pagesSpotlight Planet Magazine UpdatedOlla John IamBezaleel OluwafemiNo ratings yet
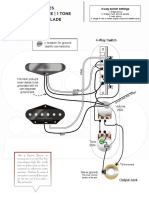
- 4-way switch wiring diagram for a 2 pickup guitarDocument1 page4-way switch wiring diagram for a 2 pickup guitarNebojša JoksimovićNo ratings yet
- Wireless DoorbellDocument20 pagesWireless Doorbellmujahed1987No ratings yet
- KSSR - MatematikDocument6 pagesKSSR - MatematikFaris FarhanNo ratings yet
- Sat - RPMSDocument4 pagesSat - RPMSAeroz RednaxelaNo ratings yet
- Chassis (LS17) PDFDocument10 pagesChassis (LS17) PDFlasky11No ratings yet
- Tablet ToolingDocument51 pagesTablet ToolingDr. Muhammad Imran Khan100% (2)
- Final ProjectDocument27 pagesFinal ProjectMohit KeshriNo ratings yet
- Reaction Paper The Flight From ConversationDocument4 pagesReaction Paper The Flight From ConversationJoe NasalitaNo ratings yet
- Differences Between Routers, Switches and HubsDocument2 pagesDifferences Between Routers, Switches and HubsPappu KhanNo ratings yet
- Diligence International Group Understands MexicoDocument2 pagesDiligence International Group Understands MexicoPR.comNo ratings yet
- Secure Email Transaction SystemDocument32 pagesSecure Email Transaction SystemGautam Sharma100% (1)
- FREE UX Books @UXlinksDocument4 pagesFREE UX Books @UXlinksSyaifudin MashuriNo ratings yet
- Swift 2002 Annual ReportDocument53 pagesSwift 2002 Annual ReportFlaviub23No ratings yet
- Safety Analysis of Mooring Hawser of FSO and SPM B PDFDocument10 pagesSafety Analysis of Mooring Hawser of FSO and SPM B PDFORUGANo ratings yet
- Precast Concrete Septic Tank 5000dsDocument1 pagePrecast Concrete Septic Tank 5000dsMarco Vega TaipeNo ratings yet
- Computed TomographyDocument94 pagesComputed TomographyBMT100% (3)
- Dekut Teaching Practice ManualDocument20 pagesDekut Teaching Practice Manualstephen njorogeNo ratings yet
- Lancashire BoilerDocument28 pagesLancashire BoilerDr. BIBIN CHIDAMBARANATHANNo ratings yet
- Bowing Styles in Irish Fiddle Playing Vol 1 - David LythDocument58 pagesBowing Styles in Irish Fiddle Playing Vol 1 - David LythEmma Harry100% (1)
- 1281 - Fire Extinguisher and Fire Safety Products Brochure S - tcm431-48382Document16 pages1281 - Fire Extinguisher and Fire Safety Products Brochure S - tcm431-48382Cv. muda karya jayaNo ratings yet
- PowerOn Fusion PDFDocument16 pagesPowerOn Fusion PDFJagan VanamaNo ratings yet
- PLAXIS Tutorial ManualDocument124 pagesPLAXIS Tutorial ManualPeteris Skels100% (2)
- 220 KV GSS, HeerapuraDocument56 pages220 KV GSS, Heerapurapikeshjain33% (3)
- Nokia 7368 ISAM ONT G-010G-A For Optical LAN Data Sheet enDocument3 pagesNokia 7368 ISAM ONT G-010G-A For Optical LAN Data Sheet enMirado AndriamihasinoroNo ratings yet
- Auditing and Electronic Data Processing (EDP)Document6 pagesAuditing and Electronic Data Processing (EDP)Lizza Marie CasidsidNo ratings yet
- Post TensioningDocument13 pagesPost TensioningAbdullah AhamedNo ratings yet
- EZSkin v3 User GuideDocument16 pagesEZSkin v3 User GuidePg1978No ratings yet
- Formalin MsdsDocument10 pagesFormalin MsdsMank WidhieNo ratings yet