You might also like
- 4AT SpanishDocument1 page4AT SpanishDavid G ParraNo ratings yet
- Análisis EstadísticoDocument9 pagesAnálisis EstadísticoFrancisco J. Cid HernándezNo ratings yet
- Historia Clínica Geríatrica SanoDocument6 pagesHistoria Clínica Geríatrica SanoFrancisco J. Cid HernándezNo ratings yet
- Fisiología CardiacaDocument3 pagesFisiología CardiacaFrancisco J. Cid HernándezNo ratings yet
- Cristian TrastornoDocument4 pagesCristian TrastornoFrancisco J. Cid HernándezNo ratings yet
- Mecanismos de Defensa en Trastornos de PersonalidadDocument3 pagesMecanismos de Defensa en Trastornos de PersonalidadFrancisco J. Cid HernándezNo ratings yet
- Resumen de Factores Sociales de DepresiónDocument2 pagesResumen de Factores Sociales de DepresiónFrancisco J. Cid HernándezNo ratings yet
- Primer Examen de GeografiaDocument4 pagesPrimer Examen de GeografiaFrancisco J. Cid HernándezNo ratings yet
- Resumen de Factores Sociales de DepresiónDocument2 pagesResumen de Factores Sociales de DepresiónFrancisco J. Cid HernándezNo ratings yet
- Primer Examen de GeografiaDocument5 pagesPrimer Examen de GeografiaFrancisco J. Cid HernándezNo ratings yet
- Preguntas Sistema VestibularDocument2 pagesPreguntas Sistema VestibularFrancisco J. Cid HernándezNo ratings yet
- Precancerosas DermatologíaDocument4 pagesPrecancerosas DermatologíaFrancisco J. Cid HernándezNo ratings yet
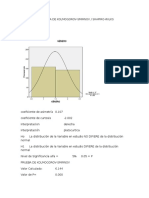
- Histogramas Kolmogrov-Shapiro Regina 19102014Document7 pagesHistogramas Kolmogrov-Shapiro Regina 19102014Francisco J. Cid HernándezNo ratings yet
- Decalogo Atención PrehospitalariaDocument4 pagesDecalogo Atención PrehospitalariaFrancisco J. Cid Hernández100% (1)
- Resumen Parte Clínica SCAsDocument10 pagesResumen Parte Clínica SCAsFrancisco J. Cid HernándezNo ratings yet
- sp131g PDFDocument5 pagessp131g PDFHermenegildo chitumbaNo ratings yet
- Resumenes AtaxiasDocument2 pagesResumenes AtaxiasFrancisco J. Cid HernándezNo ratings yet
- Diagnóstico de ObesidadDocument2 pagesDiagnóstico de ObesidadFrancisco J. Cid HernándezNo ratings yet
- Antropometria 02032016Document10 pagesAntropometria 02032016Francisco J. Cid HernándezNo ratings yet
- Circulación CoronariaDocument1 pageCirculación CoronariaFrancisco J. Cid HernándezNo ratings yet
- Analisis de VariablesDocument2 pagesAnalisis de VariablesFrancisco J. Cid HernándezNo ratings yet
- Concentrado de PacientesDocument15 pagesConcentrado de PacientesFrancisco J. Cid HernándezNo ratings yet
- Glicina 29042015Document8 pagesGlicina 29042015Francisco J. Cid HernándezNo ratings yet
- Fisiopatologia Conjuntivitis Incompleta 09022016Document17 pagesFisiopatologia Conjuntivitis Incompleta 09022016Francisco J. Cid HernándezNo ratings yet
- Dos Mujeres en La PlayaDocument1 pageDos Mujeres en La PlayaFrancisco J. Cid HernándezNo ratings yet
- Agosto-2 2015Document13 pagesAgosto-2 2015Francisco J. Cid HernándezNo ratings yet
- Arborl Casa PersonaDocument1 pageArborl Casa PersonaFrancisco J. Cid HernándezNo ratings yet
- Proyecto 1Document24 pagesProyecto 1Francisco J. Cid HernándezNo ratings yet
- Relacion de Del Personal Moras Del 1 Al 15 de Julio Del 2014Document7 pagesRelacion de Del Personal Moras Del 1 Al 15 de Julio Del 2014Francisco J. Cid HernándezNo ratings yet
- Abre Via TurasDocument1 pageAbre Via TurasFrancisco J. Cid HernándezNo ratings yet
- Análisis de texto sobre fármacos y vacunas contra el coronavirusDocument2 pagesAnálisis de texto sobre fármacos y vacunas contra el coronavirusElizabethNo ratings yet
- 806 398 SPDocument2 pages806 398 SPExiao WrNo ratings yet
- HONGOSDocument5 pagesHONGOSmarisol salgado perezNo ratings yet
- Clase Reproducción Asexual en Plantas.Document5 pagesClase Reproducción Asexual en Plantas.Julieth CamejNo ratings yet
- Taller Definición de BiologíaDocument2 pagesTaller Definición de BiologíaSantiago MartinezNo ratings yet
- Aliaga FJDocument166 pagesAliaga FJHolaqhaceNo ratings yet
- Examen Diagnòstico 3 Medio Biologìa 2018Document6 pagesExamen Diagnòstico 3 Medio Biologìa 2018jorgealfaromNo ratings yet
- Contraccion MuscularDocument39 pagesContraccion MuscularKim Hyun JoongNo ratings yet
- Diarrea Neonatal en Terneros Tesis Ucuenca Edu EcDocument95 pagesDiarrea Neonatal en Terneros Tesis Ucuenca Edu EcRoberto Ramirez0% (1)
- Cuestionario. Práctica#3Document4 pagesCuestionario. Práctica#3María Mónica DurangoNo ratings yet
- República Bolivariana de VenezuelaDocument23 pagesRepública Bolivariana de VenezuelaRodriguez SjrpNo ratings yet
- MALLA Naturales 2024Document33 pagesMALLA Naturales 2024manuel.ramirezNo ratings yet
- Quimica ForenseDocument16 pagesQuimica ForenseOreana MartinezNo ratings yet
- Impacto Social de Los Trastornos GenéticosDocument6 pagesImpacto Social de Los Trastornos GenéticosGissela MéndezNo ratings yet
- Ejercicios JoaquinaDocument14 pagesEjercicios JoaquinaAnelizNo ratings yet
- Exani I DiagnosticoDocument8 pagesExani I DiagnosticoFPOLONo ratings yet
- Fibroadenoma (Final)Document16 pagesFibroadenoma (Final)Harold Muñoz CiezaNo ratings yet
- Fisiología de Las Vías de EliminaciónDocument26 pagesFisiología de Las Vías de EliminaciónquimegiNo ratings yet
- Pseudomonas Aeruginosa PDFDocument10 pagesPseudomonas Aeruginosa PDFIsaac David Abad MatiasNo ratings yet
- Bava Final PDFDocument256 pagesBava Final PDFNatalia MoranNo ratings yet
- Anabolismo y CatavolismoDocument11 pagesAnabolismo y CatavolismotefitaNo ratings yet
- Preparación de Dna Plasmídico Por Lisis Alcalina Con SDSDocument4 pagesPreparación de Dna Plasmídico Por Lisis Alcalina Con SDSNicolas Leonardo Favi CuevasNo ratings yet
- Folleto de BiologiaDocument1 pageFolleto de BiologiaCarolina ChoezNo ratings yet
- Actualizacion en El Diagnóstico Y Tratamiento de Las Meningoencefalitis Asépticas Del PerroDocument14 pagesActualizacion en El Diagnóstico Y Tratamiento de Las Meningoencefalitis Asépticas Del PerroSamanthaRoqueNo ratings yet
- Quiz PsicobiologiaDocument5 pagesQuiz Psicobiologiacesar gomez100% (2)
- Rol de La Fibra DietariaDocument9 pagesRol de La Fibra DietariaJuliana TovarNo ratings yet
- Enzimiologia Clínica - Ensayo.Document4 pagesEnzimiologia Clínica - Ensayo.Larissa Muñoz cogolloNo ratings yet
- Historia natural de la rabiaDocument33 pagesHistoria natural de la rabiaMaicol Marin Lopez CNo ratings yet
- La MalacologíaDocument3 pagesLa MalacologíaCiber Hollerith0% (1)
- Prueba de Fuego DietmarDocument39 pagesPrueba de Fuego DietmarArnsora100% (1)