You might also like
- Instituto Nacional de Vigilancia de Medicamentos y Alimentos Luis CarlosDocument1 pageInstituto Nacional de Vigilancia de Medicamentos y Alimentos Luis CarlosCuidarenfermeria EnfermeraNo ratings yet
- Valor Medio2Document1 pageValor Medio2Cuidarenfermeria EnfermeraNo ratings yet
- Batio 33Document8 pagesBatio 33Cuidarenfermeria EnfermeraNo ratings yet
- Instrucciones de InstalaciónDocument11 pagesInstrucciones de InstalaciónAle BritoNo ratings yet
- Valor Rms de Una Señal SenoidalDocument4 pagesValor Rms de Una Señal SenoidalCuidarenfermeria EnfermeraNo ratings yet
- PoytingDocument1 pagePoytingCuidarenfermeria EnfermeraNo ratings yet
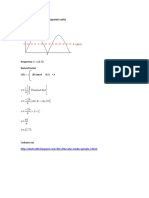
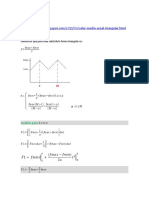
- TriangularDocument3 pagesTriangularCuidarenfermeria EnfermeraNo ratings yet
- Valor MedioDocument1 pageValor MedioCuidarenfermeria EnfermeraNo ratings yet
- Carta Tele 3 y 4Document1 pageCarta Tele 3 y 4Cuidarenfermeria EnfermeraNo ratings yet
- AUTPOSTEADOR de Taringa de VagosDocument4 pagesAUTPOSTEADOR de Taringa de VagosCuidarenfermeria EnfermeraNo ratings yet
- Valor Rms de Una SeñalDocument2 pagesValor Rms de Una SeñalCuidarenfermeria EnfermeraNo ratings yet
- Valor MedioDocument1 pageValor MedioCuidarenfermeria EnfermeraNo ratings yet
- CerezymeDocument2 pagesCerezymeCuidarenfermeria EnfermeraNo ratings yet
- Win7 SeniimpresoresDocument2 pagesWin7 SeniimpresoresCuidarenfermeria EnfermeraNo ratings yet
- Los Programas para El ComputadorDocument23 pagesLos Programas para El ComputadorCuidarenfermeria EnfermeraNo ratings yet
- EnfermedaddegaucherDocument2 pagesEnfermedaddegaucherCuidarenfermeria EnfermeraNo ratings yet
- Protejamos La NaturalezaDocument1 pageProtejamos La NaturalezaCuidarenfermeria EnfermeraNo ratings yet
- CuidadoscatetercentralDocument2 pagesCuidadoscatetercentralCuidarenfermeria EnfermeraNo ratings yet
- Descargar Libros UniversitariosDocument2 pagesDescargar Libros UniversitariosCuidarenfermeria EnfermeraNo ratings yet
- Cuidados para Paciente en QuimioterapiaDocument15 pagesCuidados para Paciente en QuimioterapiaCuidarenfermeria EnfermeraNo ratings yet
- EnfermedaddegaucherDocument2 pagesEnfermedaddegaucherCuidarenfermeria EnfermeraNo ratings yet
- RituximabDocument2 pagesRituximabCuidarenfermeria EnfermeraNo ratings yet
- Salud 2Document13 pagesSalud 2Cuidarenfermeria EnfermeraNo ratings yet
- STS Alex Primera ParteDocument137 pagesSTS Alex Primera ParteCuidarenfermeria EnfermeraNo ratings yet
- Envio de Su Querida Hermanita Margarita Noreña Desde ColombiaDocument2 pagesEnvio de Su Querida Hermanita Margarita Noreña Desde ColombiaCuidarenfermeria EnfermeraNo ratings yet
- Salud 1Document22 pagesSalud 1Cuidarenfermeria EnfermeraNo ratings yet
- Rocio. Pago Cuota Anual.Document2 pagesRocio. Pago Cuota Anual.Cuidarenfermeria EnfermeraNo ratings yet
- TRABAJODocument2 pagesTRABAJOCuidarenfermeria EnfermeraNo ratings yet
- ROCIODocument1 pageROCIOCuidarenfermeria EnfermeraNo ratings yet
- Reglas Del ClubDocument10 pagesReglas Del ClubCuidarenfermeria EnfermeraNo ratings yet
- Actualizacionesentrasplantes 2009Document862 pagesActualizacionesentrasplantes 2009Zaady Garcés Zulleymán100% (1)
- Tdah Anteproyecto PDFDocument23 pagesTdah Anteproyecto PDFDIANA MILENA PELAEZ MELONo ratings yet
- Agorafobia - Tipos y Tratamiento - MenteAmenteDocument20 pagesAgorafobia - Tipos y Tratamiento - MenteAmenteKatherine MoralesNo ratings yet
- El Cabas y El Profesional Sanitario. Cuatro Maletines Que Definen El TrabajoDocument1 pageEl Cabas y El Profesional Sanitario. Cuatro Maletines Que Definen El TrabajoYESSAMEN ESTHER COLLANTES TORRESNo ratings yet
- LEBBT03W Manual de UsuarioDocument18 pagesLEBBT03W Manual de UsuarioMijail Giovanni Medrano FuentesNo ratings yet
- Repsol Merak VDL 46Document11 pagesRepsol Merak VDL 46Juan CarlosNo ratings yet
- FM-LIR-01 y FM-LIR-02Document2 pagesFM-LIR-01 y FM-LIR-02Lucero PeñaNo ratings yet
- Maniobras ABDOMENDocument15 pagesManiobras ABDOMENANGULO REAS VICTOR ALEJANDRONo ratings yet
- Patologà A + Resumen y Preguntas de Autoevaluaciã NDocument3 pagesPatologà A + Resumen y Preguntas de Autoevaluaciã NDenisse T.C.50% (2)
- Tarea Semana 5Document7 pagesTarea Semana 5Aylin LaraNo ratings yet
- Contabilidad Especial Contabilidad de Clínicas y HospitalesDocument13 pagesContabilidad Especial Contabilidad de Clínicas y Hospitalesmax franco flores roqueNo ratings yet
- Facturación en salud: funciones clavesDocument15 pagesFacturación en salud: funciones clavesJOHN FREDY . CHITO PINONo ratings yet
- 7-Eeb-Norte-Gral-5017-0 Resistividad Del TerrenoDocument15 pages7-Eeb-Norte-Gral-5017-0 Resistividad Del TerrenoJonathan CarbajalNo ratings yet
- Eucalipto PDFDocument8 pagesEucalipto PDFomar88015No ratings yet
- Castracion QuimicaDocument3 pagesCastracion Quimicamarianela gavilanNo ratings yet
- PLANTILLA - PLAN DE PRUEBAS FUNCIONALES - AnexoDocument18 pagesPLANTILLA - PLAN DE PRUEBAS FUNCIONALES - AnexoAlvaro De La CruzNo ratings yet
- POSGI-03-ODS18 PROCEDIMIENTO INSTALACIÓN DE FAENA MEJORADO Rev 3Document28 pagesPOSGI-03-ODS18 PROCEDIMIENTO INSTALACIÓN DE FAENA MEJORADO Rev 3Luis MuñozNo ratings yet
- Evaluación de RiesgosDocument5 pagesEvaluación de RiesgosJoel Nardy Calcina BellidoNo ratings yet
- Prurito nocturno familiarDocument50 pagesPrurito nocturno familiarpatrick fils-aimeNo ratings yet
- Cuadernillo Bioseguridad 1er. CicloDocument37 pagesCuadernillo Bioseguridad 1er. CicloVero ChiuNo ratings yet
- Alimentación Saludable y Sostenida-1Document1 pageAlimentación Saludable y Sostenida-1ELVIRA REYES R.No ratings yet
- Indicadores salud niños Colombia Américas mundoDocument7 pagesIndicadores salud niños Colombia Américas mundoAmaloha RussoNo ratings yet
- Ejemplos Triada EcolDocument10 pagesEjemplos Triada EcolEloisa Cervantes100% (1)
- Consideras Que La Ley 30364 para Prevenir Erradicar y Sancionar La Violencia Contra LasDocument2 pagesConsideras Que La Ley 30364 para Prevenir Erradicar y Sancionar La Violencia Contra LasMiguel Solis TorresNo ratings yet
- Marco LegalDocument6 pagesMarco LegalSheila RamosNo ratings yet
- IT3 - Coco Instructivo TecnicoDocument20 pagesIT3 - Coco Instructivo TecnicoAlejandro Ortiz NavaNo ratings yet
- Función reproductiva y desarrollo embrionarioDocument6 pagesFunción reproductiva y desarrollo embrionariomarthadelpradoNo ratings yet
- Elaboracion de Tapabocas y Antibacterial UE TaparitoDocument20 pagesElaboracion de Tapabocas y Antibacterial UE TaparitoAcner RodriguezNo ratings yet
- Virus de La RabiaDocument23 pagesVirus de La RabiaFabricio DilasNo ratings yet
- Expo Sic Ion Humor VitreoDocument57 pagesExpo Sic Ion Humor VitreoLuna MoonNo ratings yet